


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
新型烷基甲酰吲哚类合成大麻素MDMB-4en-PINACA的检验
[代勇1  , 吴逢博
, 吴逢博2 , 吴永富3 , 周达江4 , 陆银1 , 蔡玉刚3, * , 王燕军5, * ]
, 吴逢博, 王燕军]
|
|


第一作者简介:代勇,男,重庆人,博士,教授,研究方向为毒物、毒品、微量物证检验。E-mail: Sdy-0502@163.com
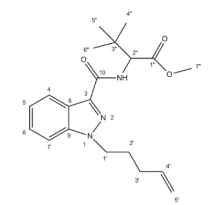
本文利用气相色谱-质谱联用(GC-MS)、红外光谱(IR)、高效液相色谱-四极杆串联飞行时间质谱(HPLC-QTOF)、核磁共振(NMR)等技术对新型烷基甲酰吲哚类合成大麻素MDMB-4en-PINACA进行结构确认。采用硅胶层析法对缴获的可疑烟叶进行纯化获得目标成分,利用GC-MS、IR、HPLC-QTOF、1H-NMR、13C-NMR等方法进行分析,确定目标物结构。结果表明,通过HPLC-QTOF获得未知化合物的精确质量数为358.2209及同位素峰簇特征,利用1H-NMR确定质子数为27及其归属,13C-NMR确定碳类型,确定分子式为C20H27N3O3,通过红外吸收确定官能团类型,确认该物质为合成大麻素MDMB-4en-PINACA。利用GC-MS、IR、HPLC-QTOF、1H-NMR、13C-NMR多种方法对未知精神活性物质进行检验具有可靠性和参考性。
The structure of MDMB-4en-PINACA (a novel indole-derived synthetic cannabinoid bearing alkylformyl groups) was here to confirm through gas chromatography-mass spectrometry (GC-MS), infrared spectroscopy (IR), high-performance liquid chromatography-quadrupole tandem time-of-flight mass spectrometry (HPLC-QTOF) and nuclear magnetic resonance (NMR). From the seized questionable tobacco leaves, the purposed analyte was extracted through silica gel chromatography, having its structure determined with GC-MS, IR, HPLC-QTOF and NMR. The accurate mass of the unknown compound (harbored with the purposed analyte) extracted from the tobacco leaves was determined as 358.2209 under HPLC-QTOF, with the isotopic cluster characteristics being defined, too. The proton number was 27 from1H-NMR and the carbon type was determined with13C-NMR, thus rendering the molecular formula as C20H27N3O3. The ascription of functional groups was verified through the related infrared absorption. The compound was accordingly confirmed to be MDMB-4en-PINACA. The combinatorial application of GC-MS, IR, HPLC-QTOF,1H-NMR and13C-NMR into identification is reliable and referential for unknown psychoactive substances.
新型精神活性物质(new psychoactive substances, NPS)在全球范围内的蔓延对社会和人类健康构成威胁。自2017年以来, 每年新增约55种NPS, 其中合成大麻素占51%以上[1]。由于部分西方国家吸食大麻合法化, 种类繁多的合成大麻素通过各种渠道流入我国。3, 3-二甲基-2-(1-(戊-4-烯-1-基)-1h-吲哚-3-羧基)丁酸酯(MDMB-4en-PINACA)是近期境外流入我国蔓延最快、发现最多的烷基甲酰吲哚类合成大麻素。
MDMB-4en-PINACA最早出现在土耳其[2, 3, 4]。目前, 国外有Ozturk等[5]对其在人体尿液中一项代谢物的研究和Watanabe等[1]利用体外肝微粒代谢对其代谢物进行分析的少量数据报道, 国内尚未见报道, 其图谱未被Mainlib、Replib、Wiley、Nist库收录, 极容易造成漏检。本文对该化合物的气相色谱-质谱联用、红外光谱、高分辨质谱和核磁谱等数据进行了研究, 为相关研究提供一定的参考。
RV10旋转蒸发仪(德国IKA公司), SHB-III型循环水式多用真空泵(郑州长城科工贸公司), SCIENTZ-12N型冷冻干燥机(宁波新芝生物科技股份公司), AV-400核磁共振仪(美国布鲁克公司), 7890B-5977B型气相色谱-质谱联用仪(美国Agilent公司), Exion-X500R超高效液相色谱-四级杆飞行时间串联质谱仪(美国ABSciex公司), 傅里叶变换红外光谱仪SpectrumFrontier(美国PerkinElmer公司), Milli-Q超纯水机(美国Millipore公司), GF254薄层层析硅胶(青岛海洋硅胶厂)。
甲醇、乙腈(色谱纯, 德国 Merck公司); 甲酸(色谱纯, 美国 Sigma-Aldrich 公司); 正己烷、乙酸乙酯、甲醇(色谱纯, 成都科隆化学品公司); 氘代甲醇(美国Cambridge Isotope Laboratories公司); 超纯水(实验室自制)。
待测检材为某吸毒现场查获的未知成分烟叶。取样品10.0 g用三氯甲烷完全浸泡后涡旋振荡2 min, 过滤提取液, 将提取液旋蒸浓缩后用正己烷溶解。用硅胶层析法纯化目标成分, 依次用3 BV(bed volume)正己烷、3 BV正己烷-乙酸乙酯(4∶ 1, 体积比)、3BV正己烷-乙酸乙酯(1∶ 1, 体积比)洗脱, 每0.5 BV收集一个馏分。以正己烷∶ 乙酸乙酯(1∶ 1, 体积比)为展开剂, 点样于GF254硅胶板, 在254 nm紫外光下观察展开结果, 对比组分, 合并相同馏分、浓缩、冻干, 得到样品约13.0 mg。
1.3.1 气相色谱-质谱联用仪条件
色谱条件:HP-5MS毛细管柱(30 m×0.25 mm×0.25 μm); 载气为高纯氦气, 进样口温度为280 ℃, 分流比为10∶ 1, 流速1 mL/min; 初温100 ℃保持2 min, 以20 ℃/min程序升温至280 ℃保持4 min, 实验用时16 min。
质谱条件:离子源温度为230 ℃; 传输线温度为250 ℃; 离子源为EI源, 电压70 ev; 溶剂延迟为2.5 min; 采用全扫描模式(Scan); 扫描范围为45~450 amu。
1.3.2 红外光谱条件
采用DTGS检测器, 3次金刚石衰减全反射(UATR)扫描。扫描范围4 000~650 cm-1, 累加扫描8次, 分辨率4 cm-1, OPD速度为0.2 cm-1/s。
1.3.3 液相色谱串联高分辨质谱条件
色谱条件:色谱柱为PhenomenexKinetex C18柱(2.1 mm×100 mm×2.6 μm); 进样量为1 μL; 流动相为0.1%甲酸水(A)-乙腈(B), 梯度洗脱模式, B相初始浓度为10%, 在1~5 min内流动相B由20%增加到95%并保持5 min; 流速为0.3 mL/min; 进样量为1 μL。
质谱条件:ESI源。正离子检测模式:离子源温度为500 ℃; 气帘气241 kPa, 碰撞气48 kPa, 雾化器345 kPa, 加热器345 kPa; 喷雾电压5.5 kV; 质量扫描范围100~1000 Da; 去簇电压70 V, 去簇电压范围0~100 V; 碰撞能范围0~50 V, 累积时间0.25 s, 碰撞能为10、20、30、40 eV。负离子检测模式:离子源温度500 ℃; 气帘气241 kPa, CAD气48 kPa, 雾化器345 kPa, 加热器345 kPa; 喷雾电压4.5 kV; 质量扫描范围100~1 000 Da; 去簇电压80V, 去簇电压范围0; 碰撞能10 V, 碰撞能范围0; 累积时间0.25 s; 碰撞能10 V。
1.3.4 核磁共振条件
溶剂为氘代甲醇, 探头温度 300.0 K, 采用标准脉冲序列。
经GC-MS分析, 被查获的烟叶提取物中检出11.809 min处质荷比为145.0、171.1、213.1、269.1、301.1、357.2等离子碎片, 检索Mainlib、Replib、Wiley14、Nist17数据库未发现匹配成分, 根据碎片特征规律推断可能为新型合成大麻素衍生物, 总离子流图及质谱图见图1, 其质谱可能裂解途径见图2。
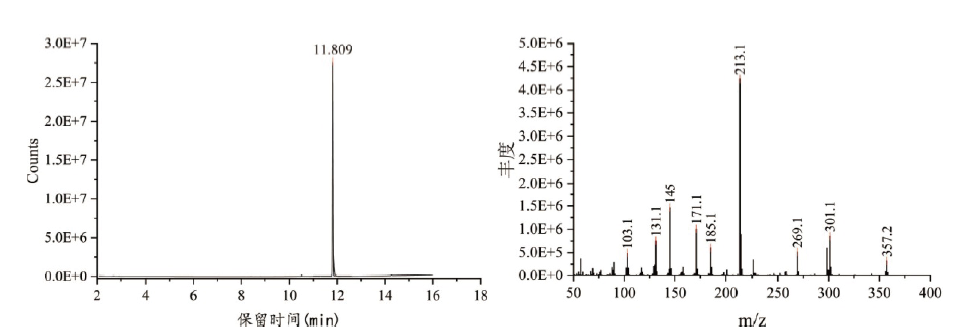 | 图1 样品总离子流图及质谱图Fig.1 Total ion-flow chromatogram and mass spectrum of the analyte extracted from sample |
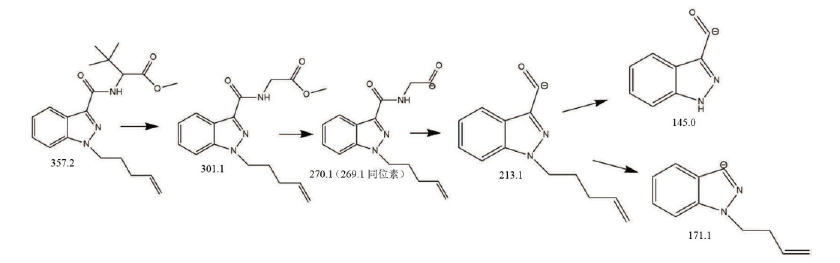 | 图2 样品气相质谱可能断裂途径Fig.2 Fragmentation pathway postulated with the GC-MS result of the analyte extracted from sample |
采用1.3.2所述方法对1.2提取的纯化物进行红外光谱分析, 如图3所示。使用Spectrumv10.5.2软件进行谱图处理, 官能团吸收峰归属如表1所示。
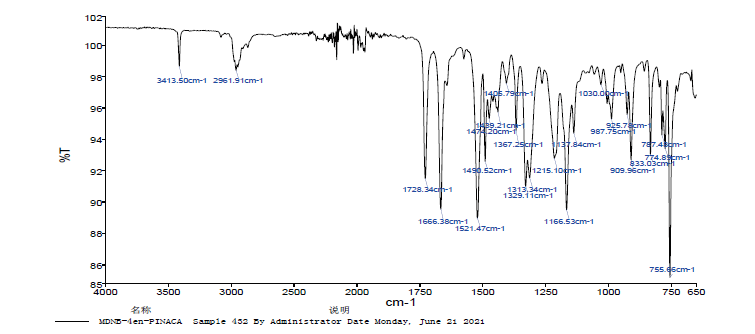 | 图3 样品红外光谱图Fig.3 Infrared spectrum of the analyte extracted from sample |
| 表1 样品红外光谱数据 Table 1 Infrared spectral data of the analyte extracted from sample |
样品高分辨质谱如图4, 其准分子离子峰为[M+H] 358.2209, 可能的碎片离子为:[M-C2H2O2-H] 298.1984、[M-C7H12O2+H] 230.1350和酰胺键断裂离子[M-C7H13NO2-H] 213.1111、[M-C12H21NO2-H] 145.0430, [M-C15H17N3O3-H] 69.0723、[M-C8H13NO3-H] 185.1131、[M-C9H15NO3-H] 171.0595、[M-C12H19NO3-H] 131.0645。根据上述精确分子量, 可知其分子式为C20H27N3O3, 偏差-0.9 ppm。样品在不同碰撞能下的二级质谱如图所示, 其主要的碎片离子为m/z 145.0430, 其二级质谱可能裂解途径如图5所示。
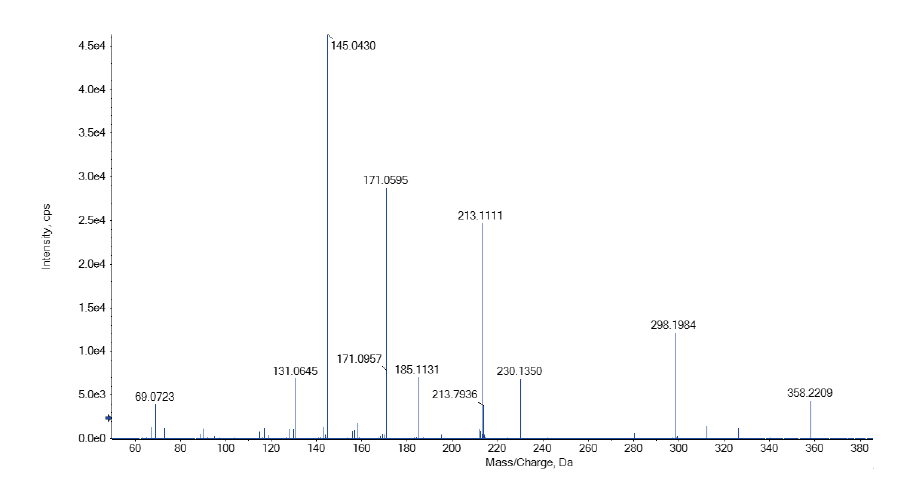 | 图4 样品高分辨质谱二级质谱Fig.4 High-resolution secondary mass spectrum of the analyte extracted from sample |
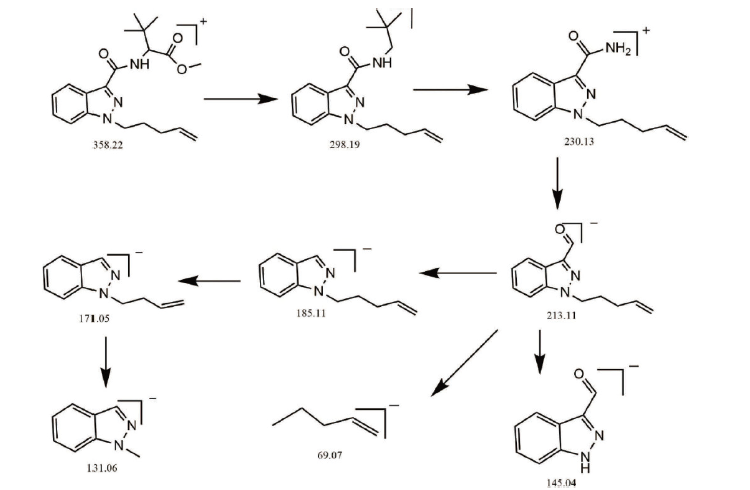 | 图5 样品的二级质谱可能断裂途径Fig.5 Fragmentation pathway postulated from secondary mass spectrum of the analyte |
2.4.1 1H核磁共振谱(1H-NMR)
经1H-NMR分析, 可见14组峰。甲基CH3:δ0.94、δ3.66 ppm单峰, 12个氢; 胺基NH:δ9.38 ppm单峰, 1个氢; 芳香环H:δ7.45、δ7.65、δ7.92、δ8.32 ppm多重峰, 4个氢; 碳碳双键H:δ4.88、δ5.13、δ5.82 ppm多重峰, 3个氢; 亚甲基CH2:δ4.46、δ1.81和δ2.16 ppm, 多重峰, 6个氢; 次甲基CH:δ4.13 ppm单峰, 1个氢, 因立体结构效应和临近N和芳香基团的影响分裂。1H-NMR数据归属见图6和表2所示。
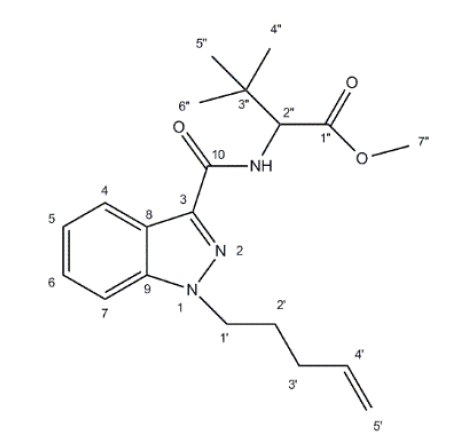 | 图6 样品的碳位命名图Fig.6 Carbon positioning into the structure of the analyte |
| 表2 样品1H-NMR数据 Table 2 1H-NMR data resulted from the tested analyte |
2.4.2 13C核磁共振谱(13C-NMR)
经13C-NMR分析, 可见18组峰:δ26.2、δ51.9 ppm为甲基碳信号; δ26.1、δ31.3、δ56.8 ppm为亚甲基碳信号; δ115.8、δ136.5 ppm为碳碳双键碳信号; δ35.9、δ120.5、δ139.7、δ142.2 ppm为季碳信号; δ160.7、δ171.5 ppm为羰基碳信号; δ66.0 ppm为次甲基碳信号; δ109.9、δ120.3、δ123.7、δ126.4 ppm为叔碳信号。13C-NMR数据归属见图6和表3所示。
| 表3 样品13C-NMR数据 Table 3 13C-NMR data resulted from the tested analyte |
样品红外光谱存在波数1728.34、1666.38、1521.49、1490.52、1217.08、987.75、909.96, 分别对应甲酯C=O伸缩振动、酰胺C=O伸缩振动、N-H弯曲振动、苯环C=C骨架伸缩振动、烷基C-N伸缩振动、=CH面外弯曲振动、=CH2面外弯曲振动, 与MDMB-4en-PINACA的结构相符。样品高分辨质谱观察到准分子离子峰[M+H] 358.2209, 对应分子式为C20H27N3O3, 与理论值偏差-0.9 ppm。此外, [C12H22NO2-H] 145.0430二级质谱信息也显示了酰胺键断裂碎片, 与MDMB-4en-PINACA的结构相符。样品1H-NMR显示了氢信号, 由1H-NMR核磁共振谱确定质子数为27, 并确定其质子归属; 样品13C-NMR显示了碳碳双键的碳信号、酰胺和甲酯的羰基碳信号的特征碳信号; 结合二级质谱和1H-NMR、13C-NMR核磁共振谱的质子和碳原子归属验证了氢-氢或碳-氢信号的相关性, 均与MDMB-4en-PINACA的结构相符。通过与文献[8, 9, 10, 11]比对和气相质谱、红外光谱、高分辨质谱和核磁共振谱分析, 可以确定样品为MDMB-4en-PINACA。
本文建立了气相色谱-质谱联用、高分辨质谱、红外光谱、1H-NMR和13C-NMR核磁共振谱技术对尚未录入谱库的未知药物MDMB-4en-PINACA的检验方法, 对质谱的数据规律进行总结, 对红外光谱官能团吸收数据、高分辨质谱精确分子量、核磁共振氢谱中质子的归属和碳谱中碳的归属进行了探索, 得到了MDMB-4en-PINACA的波谱特征数据, 为该物质的检验提供数据参考。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|