
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
SMART:自主研发的混合STR图谱分析系统
[季现超1, 2  , 池连江
, 池连江1, 2 , 徐珍3 , 彭柱3 , 叶健3 , 凃政3, * , 陈华2, * ]
, 池连江, 陈华]
|
|


第一作者简介:季现超,男,山东德州人,博士研究生,研究方向为法医遗传学。E-mail: jixianchao17d@big.ac.cn
目的 混合STR图谱的分析是法医遗传学领域的一个难题。目前国内大多数法医DNA实验室依靠人工方法分析混合STR图谱,费时费力,且拆分效果也多有主观性,难以满足日益增长的混合STR图谱分析的需求。本文介绍一套自主研发的混合STR图谱分析系统——SMART (STR Mixture Analysis and Resolution Tools),其能够实现对混合STR图谱的自动化分析,包括拆分混合STR图谱和计算似然比。方法 SMART使用的STR峰高的概率模型,考虑了影子峰、降解、基因座特异性扩增效率、峰高变异、插入峰、峰丢失等因素。模型通过比较各个基因型集合拟合当前混合STR图谱的似然值大小来推断最有可能的基因型集合,采用了马尔可夫链蒙特卡洛(MCMC)算法进行求解。结果 SMART可应用于不同试剂盒和遗传分析仪产生的数据。经使用ABI-3500XL遗传分析仪和GlobalFiler试剂盒测试,SMART能够实现对2~5人的混合STR图谱的分析,输出包括混合比例、混合图谱质量、各个贡献者单人的STR基因分型、似然比等多项结果。结论 SMART作为一款自主研发的混合STR图谱分析系统,其基本涵盖了国外同类分析系统所具有的功能,技术性能指标达到国际先进水平,能够满足一线法医工作者对于混合STR图谱分析的多样化需求,提高混合STR图谱结果的利用率。
Objective Mixed STR profiles are difficult to analyze for forensic genetics. Most domestic forensic DNA laboratories rely on manual approaches to deconvolve the mixed STR profiles, leaving into a time-consuming and laborious task from which the more or less subjective opinions result in inability to meet the ever-growing demand for analysis of mixed STR profiles. A self-developed analysis system for mixed STR profiles, SMART (STR Mixture Analysis and Resolution Tools), was to introduce here, capable of realizing automatic analysis of mixed STR profiles into their constituents through splitting. Furthermore, SMART is functional of calculating likelihood ratio to unveil whether a suspect is a contributor of the relevant mixed STR profiles.Methods SMART adopts the probabilistic model of STR peak height, taking into account of such factors as stutter, degradation, locus specific amplification efficiency, peak height variation, drop-in and drop-out. With comparison of the likelihood of each assemblage of genotypes to fit the involving mixed STR profile, SMART is to infer the most likely combination of genotypes and uses the Markov Chain Monte Carlo (MCMC) algorithm to support its inference.Results SMART has achieved applications to the data generated from different kits and genetic analyzers. As an example of utilization of ABI-3500XL genetic analyzer and GlobalFiler kit, SMART has successfully split the mixed STR profiles composed of two to five individuals, outputting multiple resultant items including mixed proportion, mixed profile quality, STR genotyping of each individual contributor and likelihood ratio.Conclusions SMART, the self-developed analysis system for mixed STR profiles, can function to cover similar foreign software systems. Its technical performance indicates that it has reached onto the international advanced level, is eligible to meet the diversified needs of frontline forensic examiners to analyze mixed STR profiles which to have consequently their utilization rate improved.
法医DNA实验室当前的主流技术是基于PCR-CE平台对DNA样本进行STR遗传标记的检测分析, 但若DNA样本包含两人或两人以上的DNA, 则会得到混合STR图谱— — 而其分析和解释是法医遗传学领域的难题[1], 其难点在于混合STR图谱受多种因素的干扰, 如影子峰、插入峰、峰丢失、峰饱和、等位基因共享、杂合不平衡、降解等, 这些因素的叠加, 就使得混合STR图谱十分复杂。
随着DNA提取和检测技术的发展, 混合DNA样本的检出比例呈逐年上升趋势, 在法庭中作为证据的应用越来越多。目前国内法医DNA实验室普遍使用人工分析[2]的方法对混合STR图谱进行解读。既往论文也报道多起通过人工拆分混合STR图谱, 成功抓获嫌疑人的案例[3, 4, 5, 6]。但随着混合DNA样本的混合组分数增多以及模板量降低, 人工分析愈益费时费力, 难以满足当前实际需求, 法医工作者亟需高效分析混合STR图谱的工具。
当前, 国际上对于混合STR图谱的分析经常借助混合STR图谱分析系统[7, 8, 9], 国内也已有几家法医DNA实验室引进了国外先进的混合STR图谱分析系统, 如STRmix。国外混合STR图谱分析系统开发较早, 经长期研究和测试完善, 在实际案件中应用效果良好, 并已得到法庭认可, 但因其购买费用昂贵, 且缺乏自主知识产权, 不适应国内应用环境, 难以大规模推广; 受国际形势影响, STRmix已于2019年末被列入对我国禁售的高科技产品清单。本文介绍一套具有完全自主知识产权的混合STR图谱分析系统SMART(STR Mixture Analysis and Resolution Tools)。通过使用该系统分析真实案例数据, 证明SMART在分析功能方面可以替代国外同类产品。SMART能够满足法医行业的科研和工作人员对于混合STR图谱分析的多种需求, 提高混合STR图谱结果的利用率。
SMART是一款自主研发的基于概率分型模型的混合STR图谱分析系统, 以JAVA语言编写, 主要功能是实现混合STR图谱的自动化分析。
SMART使用了完全连续[8, 10]的建模方法, 对混合DNA样本中各个贡献者的DNA模板量、降解水平, 基因座的特异性扩增效率, 平行扩增效率等参数进行建模, 综合考虑了影子峰、插入峰、峰丢失、峰饱和、等位基因共享、杂合不平衡等多种因素对混合STR图谱的影响。
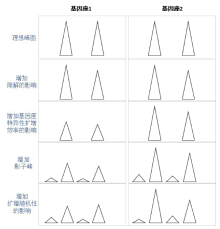
图1显示单一个体的两个杂合基因座(基因座1与2)的理想峰图为两个基因座中四个峰的高度相同, 无杂峰, 能够清晰判断分型结果, 决定峰高的因素是DNA模板量。但在实际情况中, 由于大分子的等位基因更易降解, 所以在增加降解[11]的影响后, 大分子等位基因其峰高会比小分子等位基因的低一点; 另外, 同一个试剂盒中不同基因座的特异性扩增效率[8]也不同, 反映在峰图上为基因座1的整体峰高要比基因座2的低; 且峰图中总是会有影子峰[10]存在; 最后由于扩增的随机性[12], 峰高会在一定范围内波动。这几项基本因素的叠加, 就产生了工作中常接触到的峰图。
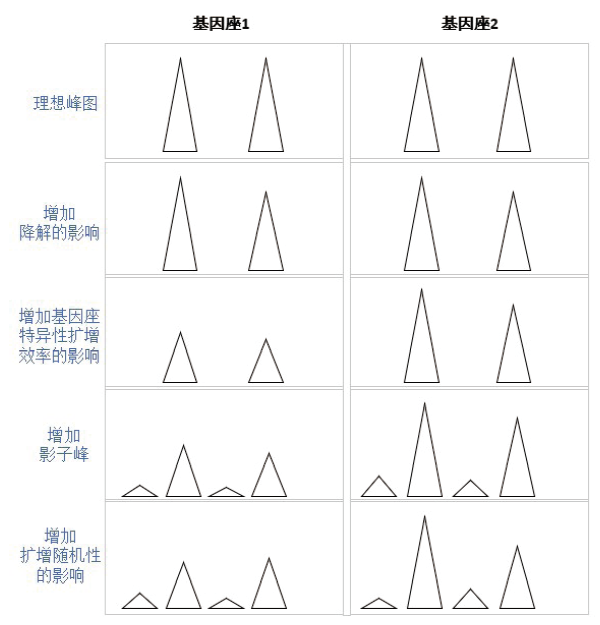 | 图1 决定峰图的基本因素Fig.1 Basic determinant factors for electrophoretogram |
将产生峰图需要的因素分为两类, 可分别进行参数化建模:第一类称为样本参数, 包括各个贡献者的基因型及其DNA模板量与降解水平, 基因座的特异性扩增效率等; 第二类称为平台参数, 包括:峰高饱和值、影子峰的比率、峰插入率、峰高随机波动的方差等因素。因样本参数无法预知, 在每次分析峰图时就需要对此进行估计。而平台参数可预先推断, 如一类试剂盒的影子峰比率可以通过统计此类试剂盒大量的单人样本的影子峰比率获知, 可在每次分析峰图时作为固定参数。
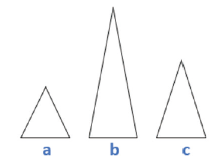
SMART所使用模型的基本假设为, 在混合STR图谱中, 不同基因座内部的各个贡献者DNA模板量的比例基本保持不变, 不同贡献者产生的峰高存在线性相加的关系。模型计算的核心是通过计算不同基因型集合产生的期望峰高与实际观测峰高拟合的似然值, 推断出各个基因型集合是真实基因型集合的概率。以一个理想的二人混合基因座说明, 如图2。峰图有三个主要的峰a、b、c, 其生成可能的候选基因型集合, 如表1。
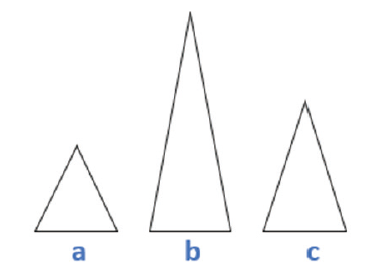 | 图2 理想的二人混合基因座Fig.2 Ideal two-person mixed locus |
| 表1 候选基因型集合 Table 1 Candidate genotype combination |
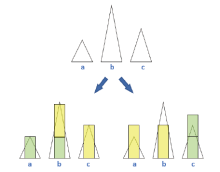
以比较(a, b ; b, c)和(c, c ; a, b)两种基因型集合的概率为例。在同一组样本参数(包括两名贡献者的DNA模板量、两名贡献者的降解水平等)下, 两组基因型集合产生的期望峰高和实际观测峰高的拟合图如图3所示, 三角形峰是观测峰, 矩形峰是期望峰, 绿色代表第一贡献者, 黄色代表第二贡献者。通过观察可看到(a, b ; b, c)产生的期望峰高与观测峰高更相近, 是真实基因型集合的概率更大。模型使用似然值来量化这种近似程度。
 | 图3 两组基因型集合的拟合图Fig.3 Fitting results from two sets of genotype combination |
通过比较不同基因型集合和不同样本参数生成的期望峰高与观测峰高拟合的似然值大小, 就可推断出贡献者基因座上各个候选基因型集合和样本参数的概率。整个计算过程需要推断的参数多达二十多个, 选用MCMC算法解决计算问题。
SMART包含六个模块, 功能分别为:
1)图谱拆分:拆分混合STR图谱, 输出分析报告, 得到混合DNA样本中各个贡献者的基因分型。
2)LR计算:计算指定个体包含在混合DNA样本中的似然比。
3)数据库搜索:提供两种模式的搜索方式, 第一种是计算数据库中每个个体包含在混合DNA样本中的似然比, 判断数据库中哪些个体可能包含在混合DNA样本中; 第二种是读取图谱拆分的结果, 使用拆分得到的图谱输入数据库比对。
4)批量处理:可以同时输入多个图谱拆分或者LR计算任务, 批量运行。
5)模型参数:训练模型需要的参数。
6)管理设置:设置软件运行的默认参数等。
混合STR图谱分析系统SMART, 可支持包括GlobalFiler扩增试剂盒和ABI-3500XL在内的多种试剂盒和遗传分析仪, 提供的功能包括:推断混合DNA样本的混合个数, 推断混合比例, 支持图谱质量和模型拟合效果的判断, 推断混合DNA样本各个贡献者基因分型和计算似然比等。
在前期研究中, SMART主要面向由GlobalFiler扩增试剂盒和ABI-3500XL遗传分析仪产生的混合STR图谱。对于不同的商业化试剂盒和遗传分析仪, 它们分型结果的准确性都已接受了严格的测试, 试剂盒和遗传分析仪间的差异主要表现为部分关键平台参数的不同, 包括峰高饱和值、影子峰比例、插入率和峰高波动的方差等参数。SMART可以使用一定量的不同试剂盒和遗传分析仪的实验室数据进行训练而得到这些关键参数, 从而可支持包括国产Typer系列试剂盒和国产GA118-16A型遗传分析仪等在内的多款产品。
目前SMART系统能够实现2~5人组成的混合STR图谱的分析。SMART在分析混合STR图谱之前, 默认要求人工输入混合人数, 并同时提供独立的功能部件, 经运行最大等位基因数量估计和极大似然估计[13]两种算法而估计出混合人数。表2给出了在模拟数据下两种估计算法的准确率, 每个混合人数下均有1 000份混合样本[13]。
| 表2 最大等位基因数量估计和极大似然估计的准确率 Table 2 The accuracy of estimated maximal allelic number and maximal likelihood |
不管是人工输入还是算法估计, 都有可能输入错误的混合人数, 即输入的混合人数小于或者大于真实的混合人数。如果输入的混合人数小于真实的混合人数, 一般是由于一个含量较少的混合DNA贡献者与含量高的混合DNA贡献者共享了较多的等位基因, 即遮盖效应严重, 此时输出结果对含量高的贡献者不会产生太大的影响, 但含量小的贡献者的信息会出现偏差甚至完全被遮盖; 如果输入的混合人数大于真实的混合人数, 一般受到影子峰、插入峰的影响, 使得某个或某几个基因座上等位基因数量偏高, 此时输出结果对含量高的贡献者不会产生太大的影响, 但对含量较少的贡献者的拆分会有影响, 此时为了解释这些多余的峰会额外输出一个不存在的贡献者基因型。
SMART的分析报告中会输出混合比例的推断结果, 可作为法医工作者人工分析混合STR图谱的参考。表3展示了一例人工配比的三个贡献者DNA含量为3:2:1的样本, DNA模板量对应数值的意义是在理想情况下三个贡献者在峰图中产生的峰高, 比例约等于3.4:2.2:1, 基本等于三个贡献者在原始样本中DNA含量的比例。但需指出的是, 由于扩增的随机性, 在有些样本中推断出的混合比例与真实的DNA含量比例会有些许差别。
| 表3 混合比例 Table 3 Mixed ratio |
如果混合DNA样本中一个贡献者占比小于10%, 混合STR图谱上就有可能丢失该贡献者的全部或部分基因型, SMART的分析报告也只能给出该贡献者的部分有效信息。这是由一代试剂盒扩增技术的局限性决定的[14], 与SMART的分析性能无关。
实际案件中, 由于DNA降解或其他因素的影响, 会使得STR图谱的质量较差甚至难以分析, 此时混合STR图谱的分析拟合效果差, 得到的结果可信度低。若提供使用者一个客观的图谱质量指标, 将有助于使用者对结果可信度作评判。
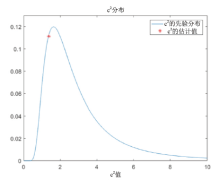
SMART使用c2作为衡量图谱质量的指标, c2是模型生成的最佳期望峰高和实际观测峰高拟合的方差大小, 服从伽马分布。c2越小说明模型生成的最佳期望峰高与观测峰高拟合的效果越好, 而当c2大于伽马分布右侧70%的分位数, 说明此时最佳期望峰高与观测峰高的差距很大, 图谱的质量很差, 该次分析的结果无效。图4展示了c2的一次估计, 估计值约为1.3, 说明图谱质量良好, 模型对数据的拟合效果好。
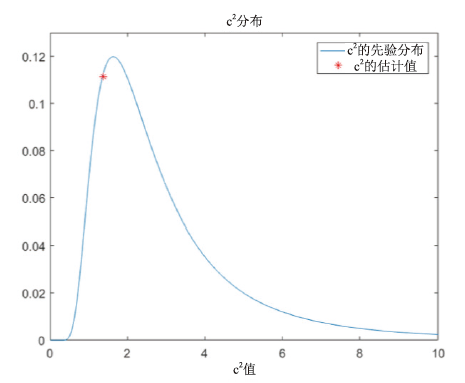 | 图4 c2分布Fig.4 c2 distribution |
本功能是SMART的核心功能之一, 是目前实际案件中混合STR图谱分析最重要的功能。SMART分两部分输出各个贡献者的基因分型。
第一, SMART给出各个贡献者在各个基因座可能的基因分型。如表4所示, 贡献者1在D16S539分型为(11, 11)的后验概率为47.5%, 分型为(12, 12)的后验概率为39.5%, 以此类推。
| 表4 贡献者1在D16S539基因座的基因分型 Table 4 Possible genotypes at locus D16S539 of contributor 1 |
第二, SMART给出了各个贡献者的最优拆分(超过预设的概率阈值)基因座集合以及对应的基因型。在设置后验概率的阈值时, 如果阈值设置太大(如99.9%), 那么获得的基因座分型的可靠性就高, 但是达到阈值的基因组数目会减少, 如果设置的阈值过低(如50%), 那么获得的基因座数目会变多, 但是达到阈值的基因分型的可靠性就变低。SMART最终汇总了各个贡献者后验概率大于90%的基因座(如表5所示)和各个贡献者后验概率大于99%的基因座(本文未给出)供人工参考。其中有的基因座如D2S441和D10S1248, 只推断出一个后验概率大于90%的等位基因, 但是不能确定另外一个等位基因(使用标记F表示, 代表当前基因座所有的等位基因)。将以上结果输出为Codis文件即可在全国DNA数据库中进行搜索, 在没有嫌疑对象的情况下可为案件侦查提供线索。
| 表5 贡献者1(51.3%)各个基因座的最优拆分(后验概率≥ 90%) Table 5 The optimal resolution obtained from splitting each locus of contributor 1 (posterior≥ 90%) |
需要指出的是, 由于表格中基因座的基因分型是在概率意义下给出的, 故仍然存在小概率出错的可能, 因此, 将拆分出来的指定个体的基因型入库比对时, 需设置等位基因容差, 防止因个别基因座的分型错误导致误排除。
本功能是SMART另外一个核心功能。目前国内大多数的报道都着重关注混合STR图谱拆分的功能, 很少关注混合STR图谱分析结果的似然比计算以及其作为法庭物证的功能。国际法医遗传学会DNA委员会[15]推荐使用似然比作为指标判断混合DNA样本是否包含嫌疑个体。似然比是在原告假设和被告假设下得到STR图谱概率的比值, 假设混合人数是N人(2≤ N≤ 5), 相关似然比的示意公式如(1)所示。

式中:H1为假设混合样本中包含嫌疑人和N-1名随机个体, H2为假设混合样本中包含了N名随机个体, O为混合STR图谱。
SMART可以直接使用图谱数据计算似然比, 也可以使用拆分阶段输出的基因型集合的后验概率计算似然比。如果似然比显著大于1, 则支持混合DNA样本包含嫌疑人, 如果似然比显著小于1, 则不支持混合DNA样本包含嫌疑人, 如果似然比接近于1, 则无法判断。
以一个实际案例数据来比较SMART和STRmix的拆分效果。本案例得到的混合STR图谱经人工研判, 结论为混合STR图谱, 主要是三个人(凶手和两名受害人)的混合, 但存在混入第四个人(无关个体)微量DNA的可能。
本案例为一起故意伤人案, 受害人为一对夫妻, 王某(丈夫)和李某(妻子), 嫌疑人为男子刘某, 作案凶器为一把单刃匕首, 混合DNA样本从匕首柄上检出。
M48磁珠法提取DNA, 使用GlobalFiler试剂盒以ProFlexTM型PCR扩增仪进行三次平行扩增, 扩增产物用ABI-3500XL遗传分析仪进行检测。经Gene-Mapper ID-X软件进行基因分型, 从图5展示的一次平行扩增的混合STR图谱中可以看出, 多个基因座的峰数量超过5个, 分析难度很大, 难以进行人工拆分。
 | 图5 案件的混合STR图谱Fig.5 Mixed STR genotype profile from a case |
两款软件的分析阈值设置为50相对荧光单位, 输入三次平行扩增得到的混合STR图谱数据文件, 受害人王某的基因分型作为已知贡献者基因型, MCMC采样次数均为燃烧期(burn-in)10万次和接受5万次。
STRmix没有推断混合人数的功能, 必须人工输入。但设置混合人数等于3人时, STRmix的计算效果不理想, 根据经验, 使用混合人数等于4人进行分析。SMART拥有推断混合人数的功能。在本案例中, 人工分析难以确定混合人数, 在运行SMART时, 选择不输入混合人数。SMART依据输入的混合STR图谱数据推断混合人数为4人。
SMART的输出报告中, 包含观测峰高和期望峰高拟合图。如图6所示, 给出了D3S1358基因座的拟合图, 包含了三次平行扩增, 红色峰是观测峰高, 蓝色峰是期望峰高。拟合图中纵坐标代表相对荧光单位的数值, 横坐标如1-15, 代表第一次平行扩增的等位基因15。从拟合图谱中, 可以直观地得到多种信息, 如期望峰高和观测峰高的拟合程度等, 有利于对软件的输出报告进行人工解读。STRmix不支持该功能。
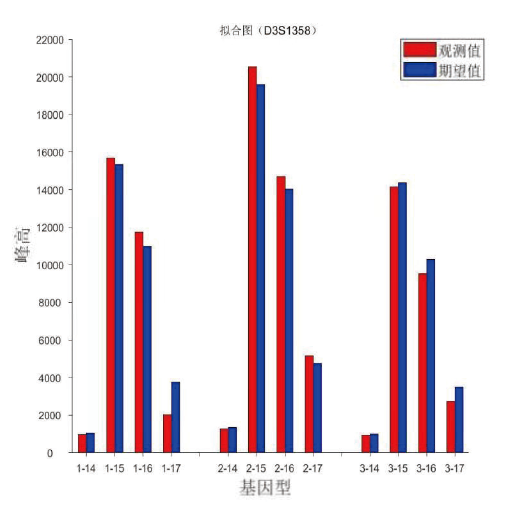 | 图6 基因座D3S1358的拟合图Fig.6 Histogram of fitting the observed peak height to the expected one from locus D3S1358 |
1)混合比例。表6显示嫌疑人男子刘某对应为第一贡献者, 受害人李某(妻子)对应为第二贡献者, 受害人王某(丈夫, 其基因分型作为混合DNA样本的已知贡献者输入到两款软件中)对应为第三贡献者, 混入的微量无关DNA为第四贡献者。两款软件的拆分比例基本一致, 在第一和第三贡献者上有差别。
| 表6 混合比例 Table 6 Mixed ratio |
2)拆分图谱。表7展示了两款软件对于第一贡献者(嫌疑人刘某)的拆分结果, 以基因分型的后验概率大于90%为标准。在21个常染色体STR基因座中, SMART推断出17个基因座完整的基因分型, 3个基因座一半的基因分型, 而同等条件下STRmix推断出14个基因座完整的基因分型, 3个基因座一半的基因分型。经和嫌疑人刘某真实基因分型比对, SMART推断的20个基因座中有19个是正确的, 而D2S1338基因座基因型(标红)拆分错误。STRmix推断的17个基因座中有13个是正确的, 而D8S1179、D10S1248、D12S391、D2S1338基因座基因型(标红)拆分错误。
| 表7 软件推断的第一贡献者(嫌疑人刘某)的分型(后验概率≥ 90%) Table 7 Genotyping of first contributor (suspect Liu) inferred by software (posterior≥ 90%) |
为了减少软件的拆分错误, 将两款软件后验概率的阈值提高到99%, 结果如表8所示。
| 表8 软件推断的第一贡献者(嫌疑人刘某)的基因分型(后验概率≥ 99%) Table 8 Genotyping of first contributor (suspect Liu) inferred by software (posterior≥ 99%) |
将基因分型的后验概率阈值提高到99%后, 21个常染色体STR基因座中, SMART推断出14个基因座完整的基因分型, 6个基因座一半的基因分型, 而同等条件下STRmix推断出9个基因座完整的基因分型, 8个基因座一半的基因分型。经和嫌疑人刘某的分型比对, SMART推断的20个基因座中有19个是正确的, 而D2S1338基因座基因型(标红)拆分错误, STRmix推断的17个基因座中有16个是正确的, D2S1338基因座基因型(标红)也拆分错误。
SMART和STRmix同属于概率分型软件, 基于相同的模型。它们的模型和计算原理已在1.1与1.2节中作简单介绍, 就是通过选取不同基因型集合和不同的样本参数生成期望峰高对混合STR图谱的观测峰高进行拟合, 最终挑选出拟合效果最好的基因型集合和样本参数。但是实际情况中, 混合STR图谱中的峰高可能会产生较大波动, 例如出现杂合不平衡, 这种情况虽然很少, 但发生后就可能会出现错误的基因分型比正确的基因分型拟合效果更好的情况, 软件就会推断出错误的基因分型, 这也是目前概率分型软件的局限所在。
SMART作为一款自主研发的混合STR图谱分析软件, 能够对混合STR图谱进行自动化分析, 输出混合人数、混合比例、混合图谱质量, 推断混合DNA样本各个贡献者的基因分型, 计算似然比。与人工分析混合STR图谱相比, 软件分析具有客观性强、速度快、拆分效果好等优势, 能够大大提高混合DNA样本物证的利用率。通过对实际案例中混合STR图谱的分析, 与国外同类型的STRmix软件相比, SMART对混合STR图谱中各贡献者分型的拆分功能已达同等水平甚至有所超越, 更符合法医行业对混合STR图谱拆分的需求。
该系统目前已完成研发与封装测试。系统的可靠性一方面需要大批量实验室样本的验证与训练, 另一方面就是大量实战数据的验证。预期该软件的推广使用将助力公安和法医学行业, 在此基础上, 有望形成混合STR图谱分析系统的业界标准。
SMART基于PCR-CE平台产生的STR图谱而开发, 使用的主要是图谱中峰的高度信息, 因而易受峰高度波动的影响。目前二代测序技术在法医遗传学领域开始崭露头角[16, 17], 二代测序技术能够检测更多的法医学位点, 包括STR、单核苷酸多态性(single nucleotide polymorphism, SNP)、微单倍型(microhaplotype)、插入缺失多态性(deletion/insertion polymorphisms, DIP)等, 能够检测更为精细化的信息, 如不仅能够检测STR的长度多态性而且能够检测STR的序列多态性。二代测序技术为解决混合DNA样本的分析问题提供了更加丰富的信息, 不过目前二代测序技术下混合DNA样本的分析尚未有系统性的研究。随着二代测序技术在行业内的推广使用, SMART会兼容混合DNA样本的二代测序数据, 更进一步地解决混合DNA样本的分析问题。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|