

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
法医学二代测序STR分型准确度与测序深度的关联性评估
[王梓齐1, 2  , 武波
, 武波2, 3 , 陈曼2 , 冯耀森2 , 张驰2 , 李明广4 , 康克莱2 , 聂胜洁1 , 王乐2, * , 吴坚1, * ]
, 武波, 吴坚]
|
|


第一作者简介:王梓齐,女,四川北川人,硕士研究生,研究方向为法医物证学。E-mail: 844093891@qq.com
目的 利用实验数据对法医学二代测序STR分型测序深度与分型结果准确度的关联性进行评估。方法 使用商业化基因组DNA制备单一来源和混合的DNA样本,以Thermo Fisher公司的25重早期测试试剂盒进行目的STR片段扩增,每种扩增产物分别使用4种不同的序列标签平行建库,并控制标记每一种序列标签的文库上机量依次占一张Ion 318芯片的1/4、1/8、1/16、1/32。经Ion PGMTM基因测序仪测序,以及Ion Torrent SuiteTM软件进行数据分析;同时对庞敬博等人发表的基于相同试剂盒和测序仪检测的95名中国汉族无关个体的6928条等位基因、影子峰和噪音序列进行测序深度统计分析,寻找测序深度与STR分型准确度的关联性。结果 各基因座测序深度随文库上样量减少而呈明显下降趋势。对于单一来源样本,每张芯片上样不超过8个均一化文库可实现全部基因座的完整分型;对于1:20比例的混合DNA,每张芯片上样不超过4个均一化文库时,未发现微量组分的等位基因丢失。人群数据测序深度统计显示,该体系基因座间存在不均衡性,有必要针对各基因座分别设定分析阈值参数。结论 测序深度与法医学STR分型结果的准确性密切相关,各基因座最低测序深度与平均测序深度的比值可作为设定分析阈值的重要参考指标。本研究确定的单张芯片上样数量仅适用于本实验体系,但相关实验设计和方案可供其他实验体系开展类似工作参考。
, WU Bo, WU Jian
Objective To assess the correlation between sequencing depth of next generation sequencing (NGS) and its resulting accuracy for forensic STR genotyping.Methods Commercial products of genomic DNA were selected to prepare single-sourced and mixed DNA samples. The targeted STR-fragment amplification was carried out with a kit of 25-plex early access STR panel from Thermo Fisher Scientific, therewith having four libraries resulted through each amplification product and different barcode adapters. The amount of libraries, linked with each barcode adapter, was controlled to respectively occupy 1/4, 1/8, 1/16 and 1/32 of one Ion 318 chip. The pooled libraries were sequenced on an Ion PGMTM machine, with the sequencing data being analyzed by way of the Ion Torrent SuiteTM software. Meanwhile, deep exploration was conducted into a dataset of 6928 sequences representing alleles, stutters and noises that were harvested from 95 unrelated Chinese Han-ethnic individuals who were the subjects of one project accomplished and reported by Pang Jingbo et al using the same kits and sequencer as this work. Thus, the NGS sequencing depth was correlatively probed with the accuracy of STR genotyping.Results The sequencing depths of each STR locus decreased significantly with the declining amounts of loaded libraries. For single-sourced samples, full genotyping profiles can be obtained when no more than 8 normalized libraries were loaded to a single chip. For mixed DNA samples at a ratio of 1:20, no dropout allele was observed from the minor contributor when no more than 4 normalized libraries were sequenced on one chip. Sequencing depth statistics showed that the co-amplification system was not balanced among STR loci, suggesting the necessity of setting an analysis threshold for each locus.Conclusions Sequencing depth closely correlates with the accuracy of forensic STR genotyping. For each locus, the ratio of minimum sequencing depth to the average can be an important indicator for setting an analysis threshold. The number of libraries loaded to a single chip depends on the available kits, machines and experimental procedures with all of which are yet applicable for the same or similar task to get reference and suggestions from this work.
测序深度一般指待测样本中某个指定核苷酸被检测的次数[1]。短串联重复序列(short tandem repeat, STR)遗传标记因其重复性, 利用二代测序技术进行检测时一般不宜对测得序列片段(read)进行拼接, 只有测通了其核心重复区的read才能被用于分型。因此, 法医学二代测序STR分型的测序深度一般使用测通的read(通常以X指代每条测通的read。本文循例也在后面相关文字处采用)数表征。测序深度是评价测序数据的重要指标之一。一般认为, 测序深度越深, 测序结果越真实可靠。二代测序技术在法医学领域的应用日趋广泛, 如何评估测序深度与法医遗传标记分型准确性的关系, 进而在物证检验中科学、合理地选择测序深度或设定数据分析中的测序深度阈值, 就成为值得研究的课题。
本文选用Thermo Fisher公司的Ion PGMTM二代测序仪和25重早期测试试剂盒进行法医STR分型实验, 并对单一来源和混合样本中微量组分与人群数据的测序深度作统计分析, 评估测序深度与分型准确度的关联性, 希冀能为法医学二代测序STR分型的测序深度选择、分析参数设定以及试剂盒和数据分析软件研发提供参考。
2800M商业化基因组DNA(Promega公司), 定量并稀释至1 ng/μ L, 作为单一来源DNA样本。9947A商业化基因组DNA(Promega公司), 定量并与2800M按照1:20的比例混合, 作为混合DNA样本。DNA定量采用Qubit® 3.0荧光计(Thermo Fisher公司)和Qubit dsDNA HS Assay Kit (500 assays)试剂盒(Thermo Fisher公司)进行。
采用25重早期测试试剂盒(Thermo Fisher 公司)对目的STR片段扩增。扩增体系为20 μ L, 单一来源DNA和混合DNA样本分别扩增4份, 每份含:8 μ L EA STR 2.5× PCR Reaction Mix v1, 2 μ L EA STR 10× PCR Primer Mix v1, 1 ng DNA模板, 用水补齐至20 μ L。扩增程序为95 ℃预变性1 min; 94 ℃、3 s, 59 ℃、16 s, 65 ℃、29 s, 进行29个循环; 4 ℃保持。
采用HID-Ion AmpliSeqTM文库试剂盒(Thermo Fisher 公司)制备测序文库。
产物孵育消化:每份20 μ L扩增产物中加入 2 μ L Fupa Reagent, 放入PCR仪中, 温控程序为50 ℃、10 min, 55 ℃、10 min, 60 ℃、20 min, 10 ℃保持。
连接序列标签:向消化后的PCR产物中加入4 μ L Switch Solution, 2 μ L序列标签预混液, 2 μ L DNA连接酶, 振荡离心后放入PCR仪中, 温控程序为:22 ℃、30 min, 72 ℃、10 min, 10 ℃、1 h。
文库纯化:使用1.5倍连接产物体积的Agencourt AMPure XP磁珠(Beckman公司)进行纯化。
文库定量:使用Ion Library TaqManTM Quantitation Kit试剂盒(Thermo Fisher 公司)和7500型荧光定量PCR仪进行。纯化后的文库稀释500倍待测。将文库标准品按照1:10梯度稀释为6.8、0.68、0.068 pM。20 μ L反应体系为:10 μ L Ion Library TaqMan qPCR Mix, 1 μ L Ion Library Taq Man Quantitation Assay, 9 μ L文库稀释液/标准品。
PCR反应程序为:50 ℃、2 min, 95 ℃预变性3 s; 变性95 ℃、3 s, 退火/延伸60 ℃、30 s, 进行40个循环。定量后将文库按照实验设计混合。
采用Ion PGMTM Hi-Q测序试剂盒(Thermo Fisher公司)以Ion PGMTM 测序仪测序, 使用Ion Torrent SuiteTM software v4.6对测序结果进行STR分型, 并从导出的Sequence histogram文件中获得每个样本测得的所有等位基因、影子峰、噪音序列及read计数信息。
对庞敬博等人(本团队早期研究项目参加成员)已报道的95名中国汉族无关个体数据[2]进行测序深度统计分析, 获取每个基因座等位基因、影子峰和噪音的最高/最低/平均测序深度。
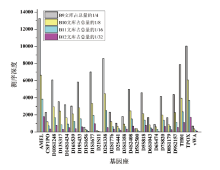
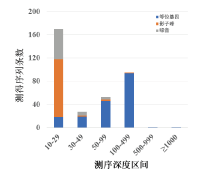
将2800M的同一扩增产物使用4种序列标签(9、10、11、12号)进行平行建库, 分别控制9、10、11、12号标签文库上机量占一张Ion 318芯片的1/4、1/8、1/16、1/32。以上文库各基因座的测序深度随文库上样量减少而下降趋势明显(图1)。
 | 图1 单一来源样本STR分型数据各基因座测序深度统计Fig.1 Performance of sequencing depths of STR genotyping into the single-sourced sample |
上样文库占318芯片1/4的总数据量为999 32X, 每个基因座的平均测序深度为3 997X; 上样文库占318芯片1/8的总数据量为52 791X, 每个基因座的平均测序深度为2 112X。其中, 9和10号标签文库出峰完整且分型完全正确, 11和12号标签文库均在D21S11基因座丢失一个等位基因, 而其余基因座分型准确, 说明使用该实验体系和Ion 318芯片进行单一来源样本的STR分型时, 一次上样8个以下(含)数量的均一化文库比较稳妥, 可以实现全部基因座完整分型; 一次上样16个以上(含)数量的均一化文库很可能导致D21S11基因座等位基因缺失而影响测序结果的准确性。
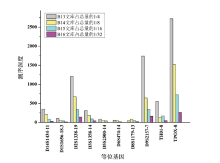
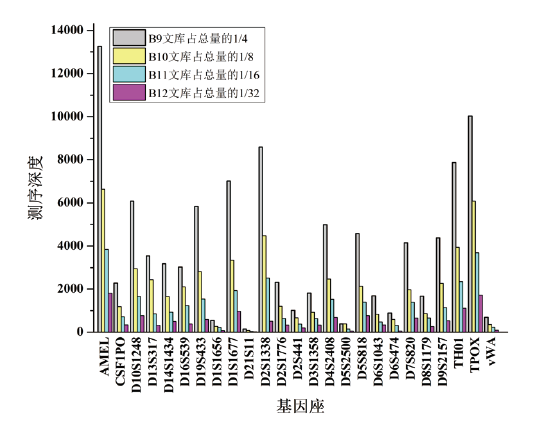
为进一步研究测序深度对混合样本检验效果的影响, 将9947A与2800M按照1:20的比例混合作为模板DNA, 采用类似于2.1的实验设计, 同一扩增产物使用4种序列标签(13、14、15、16号)平行建库, 分别控制13、14、15、16号标签文库上机量占一张Ion 318芯片的1/4、1/8、1/16、1/32。为方便观察测序深度对混合样本检验效果的影响, 剔除影子峰对数据分析的干扰, 仅分析次要组分(9947A)的10个不受影子峰干扰的等位基因, 其测序深度随文库上样量减少而下降趋势明显(图2)。
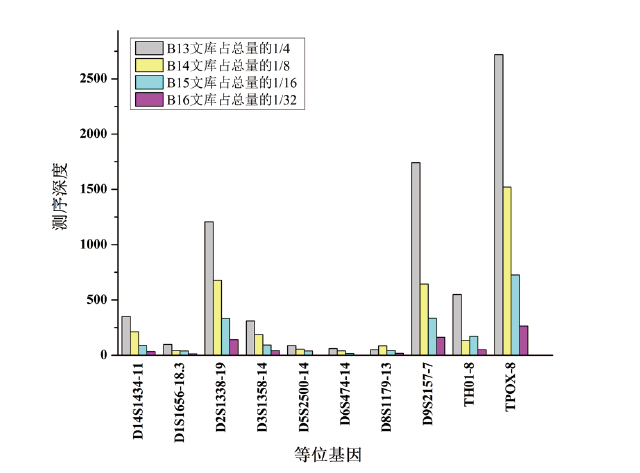 | 图2 混合样本微量组分的STR等位基因测序深度统计Fig.2 Performance of sequencing depths of the STR alleles from the minor contributors of the mixed DNA sample |
在不同文库上样量条件下, 虽然各等位基因大多可观察到一定程度的测序信号, 但若以50X作为最低测序深度阈值, 只有13号标签文库能完整报告全部10个等位基因的正确分型, 而14、15、16号标签文库只分别报告8、6、4个等位基因的正确分型。
以上结果表明, 混合DNA的正确分型对于测序深度的要求更严苛, 也说明可以通过调高测序深度的办法改善混合DNA分型效果。针对本研究采用的实验体系, 若要从1:20比例的混合物中获得次要组分的完整分型结果, 整张Ion 318芯片仅上样4个样本才比较稳妥。需要指出的是, 虽然此处关于芯片上样数的结论仅适用于本研究所采用的实验体系, 但是确定上样样本数量的实验设计和方案对于其他实验体系开展类似工作应也具有很好的参考与借鉴价值。
2019年, 本团队早期研究项目参加者庞敬博等发表了95名中国汉族无关个体的二代测序STR分型数据[2], 该文所采用的测序平台和试剂盒与本文完全相同, 95名个体的测序实验的芯片上样量分8次完成, 每张芯片的上样量在8~16个样本之间。为基于人群数据进一步分析测序深度与分型准确度的关系, 本文对该95名中国汉族无关个体数据的测序深度进行分析(见图3), 各基因座的测序深度占比如图3a所示, 等位基因、影子峰和噪音占比如图3b所示。
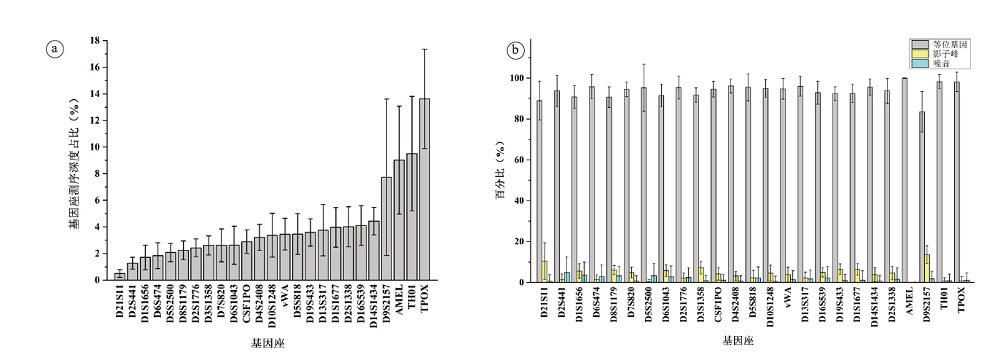 | 图3 测序深度占比(a:各基因座统计; b:各基因座中等位基因、影子峰及噪音统计)Fig.3 Percentage of sequencing depth for each locus (a) and the alleles, stutters and noises involving with each locus indicated (b) |
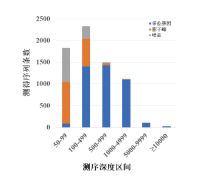
统计每个样本中测序深度≥ 50X的read, 并通过与毛细管电泳(capillary electrophoresis, CE)分型结果比对, 将每条read归类为等位基因、影子峰或噪音如表1和图4所示。结果显示, 所有≥ 5 000X的read均可归属为等位基因, 但在50X~99X、100X~499X、500X~999X、1 000X~4 999X等四个测序深度区间内, 均同时存在等位基因、影子峰和噪音序列, 且随着测序深度提高, 等位基因所占比例逐渐提高, 影子峰和噪音所占比例逐渐降低。
| 表1 95名中国汉族无关个体数据测序深度统计 Table 1 Sequencing depths and resultant genotyping presentation for the dataset of 95 unrelated Chinese Han-ethnic individuals |
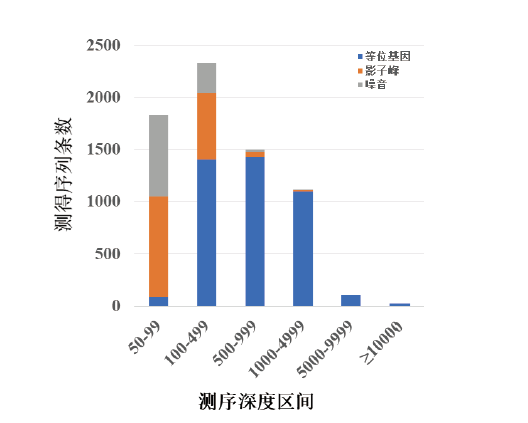 | 图4 95名中国汉族无关个体数据测序深度统计Fig.4 Genotyping componential presentation from NGS sequencing depths obtained with 95 unrelated Chinese Han-ethnic individuals |
在1 000X~4 999X测序深度区间观察到仅有一个噪音信号, 经核查, 该噪音出现在TPOX基因座(图5), 该样本在TPOX基因座为纯合子, 分型为9, 测序深度7 445X; 而这个特殊的噪音信号出现在影子峰(TPOX-8)位置, 测序深度达1 557X。在进行数据统计时, 本研究对影子峰的界定条件为:出现在影子峰位置且测序深度低于所对应等位基因测序深度的20%。该噪音信号的测序深度占TPOX-9等位基因测序深度的20.91%, 所以未被计入影子峰, 但经过人工核查, 该信号可能是TPOX-9的影子峰。
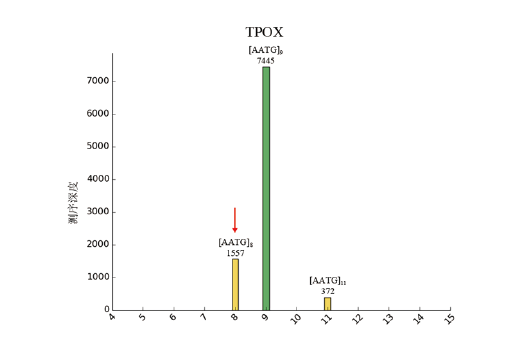 | 图5 95名汉族无关个体数据中测序深度最高的噪音Fig.5 Noise from TPOX locus which to yet attain the highest sequencing depth with the dataset of 95 unrelated Chinese Han-ethnic individuals |
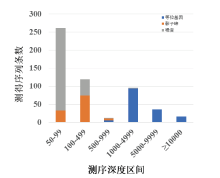
基于以上统计结果, 难以实现依据测序深度将等位基因与影子峰、噪音做出区分, 主要原因之一是该实验体系各基因座间均衡性并非理想(图3)。因此, 按照以上的数据分析思路, 对该体系中平均测序深度最高和最低的基因座作进一步分析, 即TPOX基因和D21S11基因座。如表2和图6所示, TPOX基因座499X以下测序深度的所有测得序列均为影子峰或噪音, 1 000X以上测序深度的所有测得序列均为等位基因, 在500X~999X区间段等位基因、影子峰和噪音都存在。虽然无法做到完全区分, 但可为数据分析提供重要参考, 相比于将所有基因座一起统计的数据(表1)其区分效果好得多。
| 表2 TPOX和D21S11基因座测序深度统计 Table 2 Sequencing depths and resultant genotyping presentation for the loci of TPOX and D21S11 |
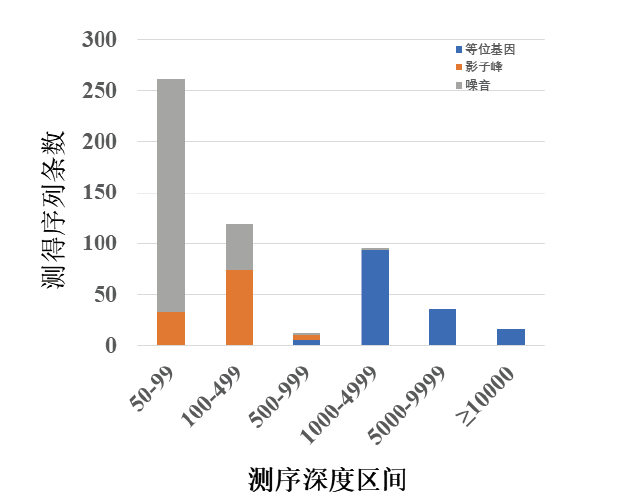 | 图6 TPOX基因座测序深度统计Fig.6 Genotyping componential presentation from NGS sequencing depths with the TPOX locus |
由于D21S11基因座整体测序深度较浅, 有些等位基因的测序深度甚至不足50X, 如果继续按照以上的测序深度区段进行统计会造成等位基因丢失, 所以增加设置了10X~29X和30X~49X两个测序深度区间进行统计。结果表明, D21S11基因座10X~29X测序深度区段的测得序列中89%为影子峰或噪音, 30X以上测序深度的测得序列91%为等位基因(表2和图7)。这样处理虽然无法做到完全区分, 但显然好于将所有基因座一起统计的数据区分效果。
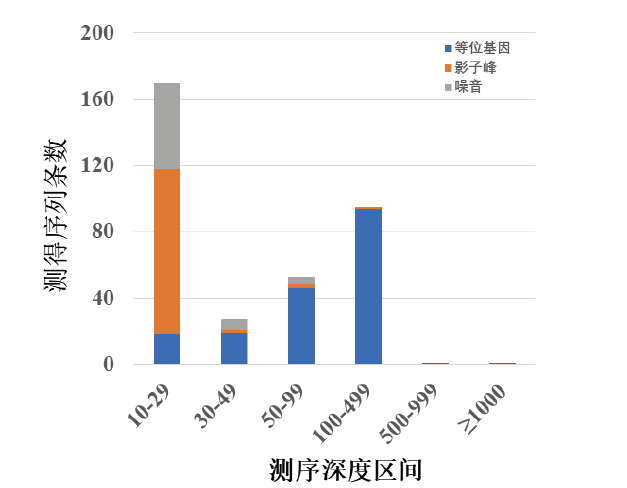 | 图7 D21S11基因座测序深度统计Fig.7 Genotyping componential presentation from NGS sequencing depths about the D21S11 locus |
以上结果表明, 复合STR分型体系中各基因座间测序深度的均衡性十分重要。基因座间的不均衡性很可能导致无法采用统一的测序深度分析阈值。这种情况下, 有必要针对各基因座设定各自的测序深度阈值。但是, 经进一步统计95名汉族个体数据中各基因座等位基因的最高、最低和平均测序深度以及影子峰和噪音的最高测序深度(表3), 可知各基因座影子峰和噪音的最高测序深度基本都高于等位基因的最低测序深度, 因此无法通过设定阈值进行彻底区分。不过, 计算求得各基因座最低测序深度与平均测序深度的比值基本在0.1以上, 例外的是D9S2157、D2S1338、D19S433三个基因座的最低测序深度与平均测序深度比值分别为0.027 6、0.054 5和0.059 3。故综合以上数据应可为分析阈值的设定和结果判定提供重要参考。
| 表3 95名中国汉族无关个体各基因座测序深度统计 Table 3 Sequencing depths and resultant genotyping presentation for each locus from 95 unrelated Chinese Han-ethnic individuals |
相比毛细管电泳技术, 基于二代测序进行STR分型能更进一步揭示序列的多态性信息, 展示STR等位基因重复单元和侧翼序列的真实差异, 从而实现更加精细化的STR分型[3, 4, 5], 尤其在检测高度降解样本和区分混合样本方面会具有优势[6]。
在统计测序深度的过程中, 95名汉族个体数据中的D21S11、D3S1358、D2S441、D8S1179、D4S2408、D2S1338、D5S2500、vWA、D19S433、D6S474等10个基因座其毛细管电泳检测表现为纯合子而二代测序则为杂合子(即等长杂合子)。
表4中, 各基因座报告的等长杂合子数量占CE纯合子数量的比例在20%左右, 占样本总数比例在4%左右。其中, 在D21S11基因座观察到的等长杂合子占CE纯合子的比例最高, 达31.58%, 占样本总数比例达6.32%。
| 表4 各基因座等长杂合子统计 Table 4 Isometric heterozygotes of the STR loci observed with NGS |
以上结果与Borsting等[7]的报道相似, 他们利用二代测序技术对PCR-CE平台检测结果进行检验, 发现30%的纯合子呈现为杂合子。由此可见, 二代测序技术揭示的等长杂合子情况出现概率较高, 显示二代测序技术能够实现更精细化STR分型, 可为法医混合DNA分型拆分提供更多的支撑作用[8, 9, 10]。
本文基于Ion PGMTM二代测序系统和25重早期测试试剂盒开展测序深度对STR分型结果准确性的影响分析。测序深度与测序芯片通量、测序样本数、检测基因座数目、基因座间均衡性、测序数据质量等因素紧密相关。刘明艳等[11]研究了16S rRNA基因测序深度对油藏细菌菌群分析的影响, 表明增加测序深度可以检测到样本中极低丰度的微生物类群。这与何世耀等[12]研究结论一致, 测序深度的不同会对测序数据产生显著影响。这种影响主要体现在较深的测序深度会显著增加稀有微生物的丰富度, 进而有利于增强对环境微生物群落整体功能的认识。Jung等[13]研究结果表明, 足够的测序深度和适当的峰值调用算法对于确保染色质免疫沉淀后高通量测序(ChIP-seq)数据所得结论的稳健性至关重要。
以上研究表明测序深度对二代测序数据分析准确性尤为重要。本文将同一扩增产物标记4种不同的序列标签, 再通过控制文库上样量实现对样本测序深度的差异化设定, 这种实验设计与通常测序前均一化文库的方法不同[14, 15]。
在本文实验体系中, 文库各基因座的测序深度都随文库上样量减少而呈明显下降趋势。单一来源样本芯片占比为1/4、1/8、1/16、1/32时其分型准确率分别为100%、100%、97.6%、97.6%。
混合DNA是法医物证检验的技术难题, 虽然国内外专家从多方面开展了大量科研工作, 但都很难实现完满解决。相比毛细管电泳技术, 二代测序STR可报告等长杂合子等精细化信息, 能辅助混合DNA的分型拆分。一般认为毛细管电泳技术可从1:8比例的混合样本中获得微量组分的完整分型, 而1:20的混合样本则超出毛细管电泳的检测能力。
本研究探索性地检测了1:20比例的混合DNA, 混合样本芯片占比为1/4、1/8、1/16、1/32时其分型检出率分别为100%、80%、60%、40%。另外, 95名汉族个体的数据有10个基因座观察到等长杂合子现象, 占CE纯合子的比例平均达18.94%, 故二代测序STR可辅助混合DNA分析进行量化评估。
人群数据测序深度统计显示, 所选用体系的基因座间存在不均衡性, 故有必要针对各基因座分别设定分析阈值参数。对运用法医学二代测序进行案件样本检验的实验室, 本文应具有参考价值, 如实验设计可针对所选用的测序平台、芯片、试剂体系开展内部确证研究, 以积累基础数据, 确定检测单一来源样本、混合样本时每张芯片的上样数以及各基因座的分析阈值。
本文还发现, 统计各基因座的最高、最低和平均测序深度, 并将最低测序深度与平均测序深度的比值作为设定分析阈值的重要参考指标, 也有助益。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|