

{kind=link}
{kind=link}
{kind=link}
{kind=link}
构建精液特异的DNA甲基化-SNP分型体系并试测精液来源
[谢博文 , 宋凤, 罗海玻, 王双双, 张珂, 李英碧
, 宋凤, 罗海玻, 王双双, 张珂, 李英碧* ]
, 宋凤, 罗海玻, 王双双, 张珂, 李英碧]
|
|


* 通讯作者简介:李英碧,男,云南昆明人,硕士,教授,研究方向为法医物证学。E-mail: liyingbiscu@163.com
第一作者简介:谢博文,男,湖南邵阳人,硕士在读,研究方向为法医物证学。E-mail: 2532230980 @qq.com
目的 初步构建DNA甲基化联合SNP分型体系,探索其用于精液来源鉴定的价值。方法 首先通过分析甲基化芯片数据筛选出精液特异性的甲基化位点,通过软件设计出各位点特异性引物,再利用SNaPshot技术对各位点相邻的SNP进行检测,尝试鉴定精液来源。结果 本实验成功构建了以精液作为鉴定对象的四位点甲基化-SNP分型复合检测体系,其对62份随机取样的中国汉族体液样本的鉴定结果表明其特异性良好,可准确区分出44份精液或精斑样本与其他三类共18份体液样本。且其灵敏度佳,在转化DNA量为0.5ng时仍可获得完整分型。在混合斑实验中,当精液与阴道分泌液体积比例为1∶40时,该复合体系仍能获得精液完整的分型图谱。结论 该四位点复合分型体系在精液鉴定中具有重要价值,证明了DNA甲基化联合SNP分型体系用于体液鉴定的可行性。
Objective To trial-construct the methylated-DNA-orienting multiplex SNP typing system and explore its value in identifying human semen samples.Methods The human semen-specific methylation sites were selected through analyzing the data from DNA methylation microarray. Specific primers of the selected sites were afterwards designed with several suites of pertinent software. SNaPshot was used to detect the neighboring single nucleotide polymorphisms (SNPs), thereby helping make the human semen origin able to be deduced.Results The 4-plex DNA methylated-locus-orienting SNP typing system for identifying human semen was successfully constructed, leading that 62 body fluids randomly collected from Chinese Han-ethnic individuals were showed of desirable human semen-specificity. The system has therefore accurately differentiated 44 human semen samples from the other 18 samples of three kinds of non-semen origin. Meanwhile, Unambiguous profiles can be even exposed when the bisulfite-converted DNA was input of only 0.5ng. Satisfactory typing profiles were indeed presented out of the mixed stains that were resulted from mixing human semen and vaginal fluid in volume ratio of 1:40.Conclusions This 4-plex SNP typing system is of great value in identifying human semen. Besides, it proves that the DNA methylated-locus-orienting multiplex SNP typing system is practical for identifying human body fluids.
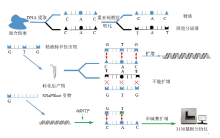
犯罪现场未知斑痕的来源鉴定可提供关于犯罪嫌疑人及其犯罪行为的大量信息, 对于重建犯罪现场, 推断犯罪性质具有重要意义[1]。然而, 由于常规检测方法存在某些缺陷, 使得一些现场检材即便进行了检测也不能得到准确而完整的分型信息。如使用镜检法鉴定无精嫌疑人的精斑或利用PSA检测试剂检测高度降解的精斑样本, 常会出现假阴性结果[1]。故研究者们开始寻求分子生物学标记物以进行体液鉴定, 其中研究较多的主要有mRNA及miRNA、DNA甲基化和微生物16S rRNA[2, 3]。mRNA及miRNA具有较强的体液特异性及检测灵敏度, 且其可进行复合扩增, 能够同时分析多种体液类型, 但它们不稳定、易降解的缺点又限制了其在法医实践中的运用[4]。相较于mRNA及miRNA, 具有体液特异性的微生物16S rRNA稳定性更高, 但现阶段其还只能用于唾液及阴道分泌液的检测, 无法对血液和精液做出准确的判断[5]。随着表观遗传学的迅速发展, DNA甲基化开始成为法医体液鉴定的热点。因其性质稳定, 不易降解, 可兼容于常规法医鉴定的STR分型平台, 同时其又可用于陈旧检材的检验, 对于侦破冷案有一定的价值[6, 7]。运用DNA甲基化进行体液鉴定需要比较不同体液在特定甲基化区域的甲基化程度值。最经典的研究方法是利用呈甲基化敏感性的限制性内切酶对甲基化区不切割的特性, 将DNA消化为不同大小的片段后再进行分析[2, 8, 9]。但其只能对特定位点进行分析, 且对样品需求量也大。目前法医学研究中常用的为DNA模板经重亚硫酸盐转化后的焦磷酸测序或单核苷酸延伸测序(SNaPshot)法。焦磷酸测序通量高, 可用于CpG位点定量分析。单核苷酸延伸测序则可同时进行不同位点CpG甲基化的测量[10, 11, 12, 13]。随着全基因组甲基化谱芯片技术的不断完善, 越来越多不同体液的特异性甲基化位点被发掘出来[14, 15, 16, 17]。然而, 对于犯罪现场斑痕的来源鉴定不仅需要判断其种类, 更需要获得其分型信息以进行个人识别才更有意义和价值。同时在法医学实践中, 斑痕多以混合斑的形式呈现, 在高度不均衡的混合样本中, 常规STR技术往往无法进行准确分型[18]。本研究尝试通过设计针对精液样本的特异性扩增引物, 从而仅使精液样本的DNA得到扩增, 排除其他体液DNA的影响; 再通过设计特定甲基化位点相邻SNP的单碱基测序引物, 结合SNaPshot技术及毛细管电泳技术以获得精液在不同位点的分型信息而最终实现其准确的来源鉴定(图1)。
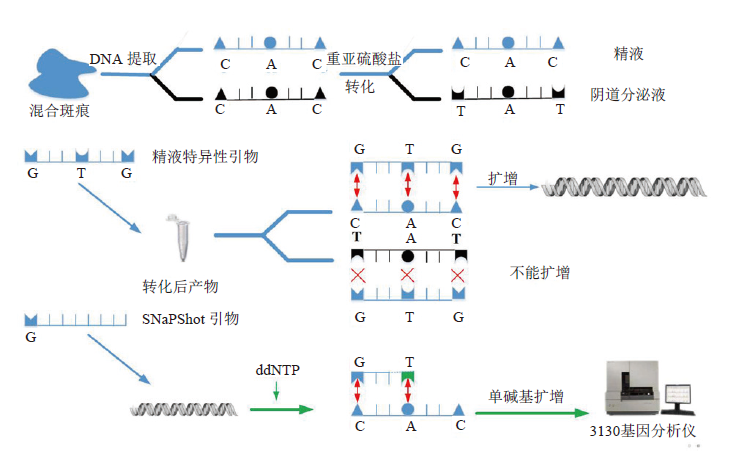 | 图1 基于DNA甲基化的精液SNP分型技术的主要原理 Fig.1 The principle of DNA methylated-locus-orienting multiplex SNP typing system |
为筛选出具有精液特异性的甲基化位点, 首先从美国国家生物信息中心 (National Center for Biotechnology Information, NCBI)的基因表达共享集 (Gene Expression Omnibus, GEO)数据库中获取了名为GSE59509的甲基化芯片数据文件。其包含了精液、唾液、外周血、阴道分泌液及月经血等各种体液的475 192个甲基化位点的相关信息。位点筛选标准如下:1)位点在精液和非精液样本中的甲基化程度值差异应大于0.8; 2)位点相邻SNP的最小基因频率(MAF)应小于0.5, 大于0.2。
实验引物设计主要分为两部分:第一为精液特异性扩增引物的设计, 该步利用甲基化位点扩增引物设计软件Methprimer进行设计。第二为单碱基测序引物的设计, 主要借助PyroMark Assay Design Software 2.0进行设计。各位点引物的具体信息见表1。
| 表1 各位点特异性扩增引物及测序引物 Table 1 Locus-specific primers of both amplification and sequencing for methylation-orienting semen detection |
根据知情同意原则, 采集62份中国汉族无关个体的体液样本:38份精液、6份陈旧精斑、6份口腔擦拭子、6份阴道分泌液及其擦拭子、6份外周血。从中随机选取一份精液样本及一份阴道分泌液样本制作混合斑, 将不同体积比例的精液和阴道分泌液进行充分混合后置于棉布上, 干燥后于室温下保存。取材时, 各种检材的取材量分别为:200 μ L血液, 100 μ L精液, 1 cm2斑痕物(陈旧精斑、口腔擦拭子、阴道擦拭子及混合斑样本)。本文所取用样本及检材均经四川大学医学伦理委员会审查通过。
DNA提取按照QIAamp DNA Mini试剂盒(德国, QIAGEN公司)的操作说明进行; 精液DNA的提取还需额外添加DTT以促进精子细胞核膜的溶解而释放出DNA。最后使用 NanoDrop 2000c分光光度计(美国, Thermo Scientific公司)对DNA浓度进行定量。
取1 μ g全基因组DNA, 按照EpiTect Bisulfite试剂盒(德国, QIAGEN公司)说明书对提取的DNA进行重亚硫酸盐转化。转化时需同时设置两组对照(完全甲基化和完全未甲基化的DNA)。转化后的DNA使用NanoDrop 2000c分光光度计进行定量后, 保存在-20 ℃的冰箱中。
1)SNaPshot测序前的特异性扩增。PCR扩增体系为5 μ L:1 μ L重亚硫酸盐转化后DNA, 0.5 μ L不含RNA酶的水, 2.5 μ L 1× PyroMark PCR Master Mix (德国, QIAGEN公司), 1 μ L复合引物。PCR热循环参数为:94℃、 11 min; 94 ℃、20 s, 60 ℃、1 min, 72 ℃、30 s, 30个循环; 72 ℃、7 min。2)扩增反应完成后, 添加虾碱性磷酸酶(shrimp alkaline phosphatase, SAP)和核酸外切酶I (Exonuclease I, EXO- I)对产物纯化, 消除多余的引物和核苷酸。3)纯化完成后开始进行单碱基延伸测序反应, 其反应体系为5 μ L:2 μ L纯化后DNA, 0.5 μ L不含RNA酶的水, 1.5 μ L SNaPshot Multiplex Ready Reaction Mix, 1 μ L复合引物。热循环参数为:96 ℃、10 s; 50 ℃、5 s, 60 ℃、30 s, 72 ℃、30 s, 26个循环。反应完成后再添加SAP消化未结合的dd NTP。最后使用ABI PRISM 3130基因分析仪对测序结果进行分析(为了使分析结果更加准确, 只对100 RFU以上的峰进行分析)。
在对甲基化芯片数据GSE59509进行分析后, 共挑选出了41个符合要求的位点(表2)。进行引物设计时, 有14个位点因无法设计出符合要求的精液特异性引物而排除。为了对剩下位点的精液特异性进行初步验证, 使用聚丙烯酰胺凝胶电泳将各位点在精液与非精液样本中进行初步扩增后的产物进行分析, 其中有10个位点未出现明显条带, 另4个位点除目的条带外, 还扩增了较多的杂带, 另有4个位点在非精液样本中也出现了目的条带, 只有9个位点在精液样本中呈现出单一而均匀的条带。最后综合考虑各位点在染色体上的分布、SNP分型等问题, 选择了其中4个位点(表3)进行复合体系的构建。
| 表2 备选甲基化位点筛选情况 Table 2 Screening of the candidate methylated loci |
| 表3 复合体系各位点及相邻SNP信息 Table 3 Each selected methylated-locus and the adjacent SNPs |
在进行复合体系构建前, 需要对各个位点进行单独扩增以验证位点用于SNP分型的可行性。为了保证实验的准确性, 随机取用5份转化后的精液DNA用于各位点的验证。毛细管电泳结果显示:各个精液样本在每个位点均出现了目标峰, 各个峰的高度均在2 000 RFU以上, 且无明显杂峰出现。
将验证了其SNP分型可行性的位点用于构建复合体系。为了使各位点均能得到较好的扩增效果, 各位点引物的比例进行了调整。如图2, 经SNP分型复合体系, 可同时得到一份精液样本中四个SNP位点的分型信息, 5份样本中各位点峰高较单扩时均有所降低。为了证实该复合体系的组织特异性, 将提取的其他种类体液DNA转化后再等量混合也做鉴定实验, 结果表明含外周血、阴道擦拭子及口腔拭子的转化样本混合物均未呈现明显条带。
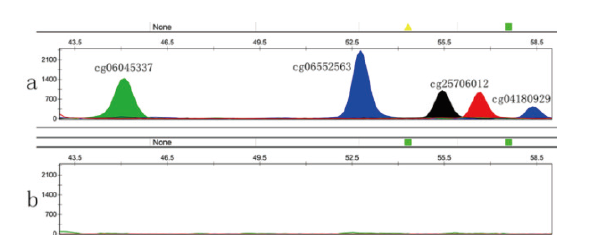 | 图2 精液样本及其他种类体液DNA通过复合体系检测后的分型图(a:精液; b:外周血、阴道擦拭子及口腔拭子的转化样本混合物) Fig.2 The 4-plex SNP multiplex typing profiles of either semen or mixture from three body fluids (a: semen; b: the mixture of bisulfite-converted DNA with venous blood, vaginal fluid and oral swabs) |
在复合体系构建好后, 对另外33份精液样本也进行了鉴定。毛细管电泳结果显示所有样本在四个位点上均具有分型信息。然而, 在对分型信息进行统计后, 发现位点cg06045337、cg06552563在所有样本中分别只出现了分型T、G。位点cg25706012在23份样本中都出现了两种分型, 另外有6份为分型G, 9份为分型A。位点cg04180929在31份样本中都呈现为分型G。其他7份样本在cg04180929的分型为:3份样本为AG, 四份样本为A。
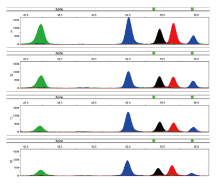
分别使用5.0、2.5、1.0、0.5 ng转化后的精液DNA对该体系的灵敏度进行评估。如图3, 在加入的DNA只有0.5 ng时, 虽然各位点峰高均降至1 000 RFU以下, 但仍可得到具有完整分型信息的电泳峰图。为了测试该体系对于法医物证精液斑鉴定的价值, 选6份精斑样本进行了鉴定。6份样本中, 除第6份在位点cg04180929处无明显峰图出现, 其他样本均呈现完整峰型(表4)。在对该样本重新进行DNA提取转化后, 又成功获得了其在cg04180929的分型。
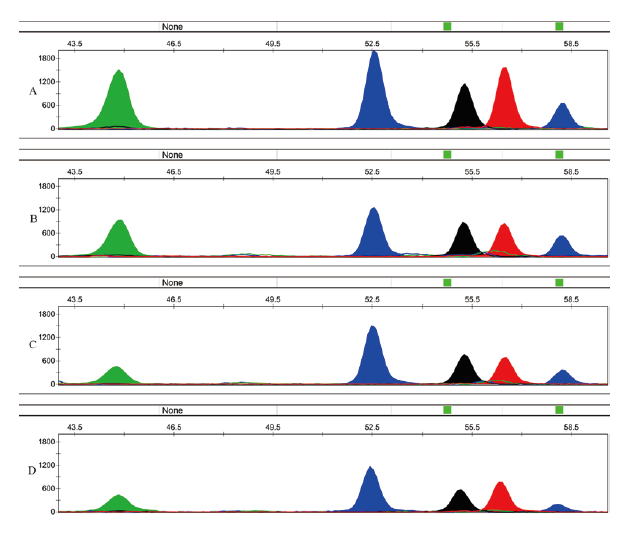 | 图3 复合体系在加入不同量精液DNA时的分型结果(A、B、C、D分别为5、2.5、1、0.5ng) Fig.3 The typing profiles of input-amount-different seminal DNA amplified by the 4-plex SNP multiplex system (A, B, C, D respectively stands for the DNA quantity of 5, 2.5, 1, 0.5ng) |
| 表4 6份精斑样本的分型信息 Table 4 Typing information of the selected six semen stains |
为了探索复合体系检测混合斑的效能, 对不同体积比的精液和阴道分泌液(精液∶ 阴道分泌液分别为1∶ 5、1∶ 10、1∶ 15、1∶ 20、1∶ 30、1∶ 40及1∶ 50)所制作的混合斑进行检测。如图4, 在混合斑比例为1∶ 50时, 位点cg06552563和cg25706012仍具有明显峰形, 峰高在800 RFU左右。但位点cg06045337和cg04180929峰图已不明显, 均低于100 RFU。
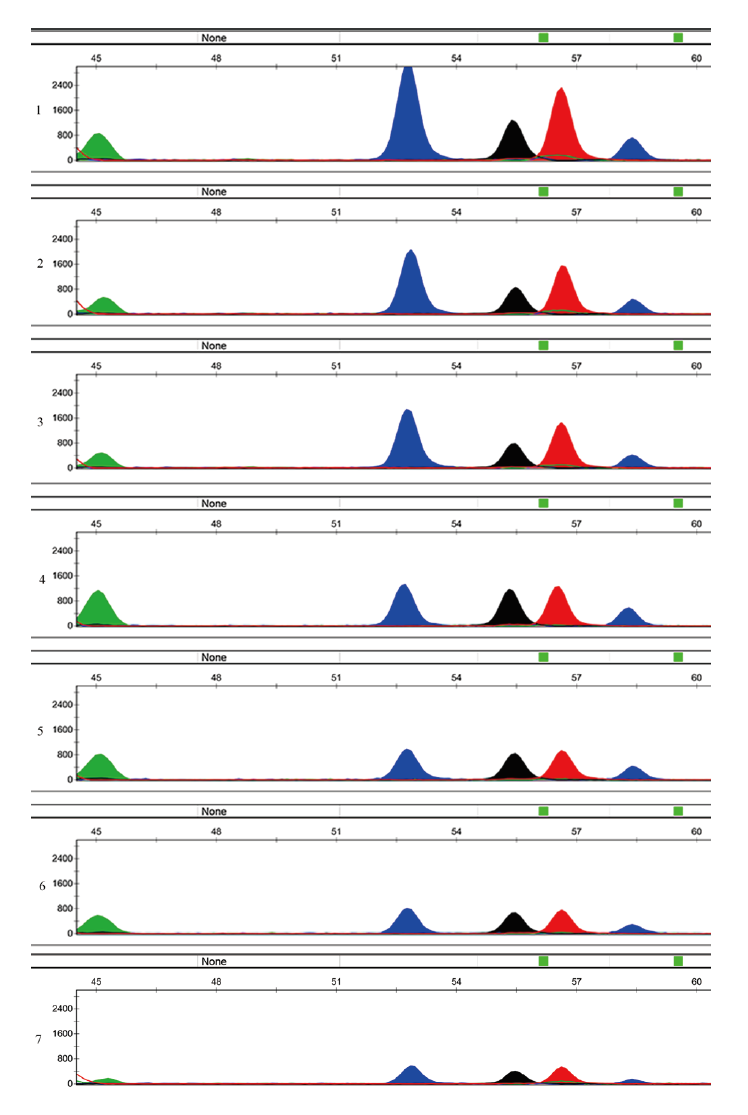 | 图4 复合体系在不同比例的混合斑中的分型结果(1、2、3、4、5、6、7的精液与阴道分泌液的混合比分别为1∶ 5、1∶ 10、1∶ 15、1∶ 20、1∶ 30、1∶ 40 及1∶ 50) Fig.4 The profiles obtained with the 4-plex SNP multiplexing system to identify the semen-and-vaginal-fluid mixture made with different-volume ratio (1, 2, 3, 4, 5, 6, 7 respectively stands for the ratio of 1∶ 5, 1∶ 10, 1∶ 15, 1∶ 20, 1∶ 30, 1∶ 40, 1∶ 50) |
体液鉴定在法医学领域一直备受关注, 研究者们对此也提出了许多方法。DNA甲基化以其独有优势正成为该研究领域的热点分子标记物。目前, 针对甲基化的大多数研究都只是从体液斑的种类鉴定出发, 不能解决常见的实际混合斑问题[14, 15, 19]。本研究基于甲基化芯片的分析数据, 结合SNaPshot技术, 以特异性甲基化位点邻近的SNP位点为检测指标, 初步构建了用于精液来源鉴定的复合体系。在体系构建过程中, 最关键的步骤是要找出具有高精液特异性的位点。在本研究中, 进行数据初筛后选出的23个位点中, 有5个点特异性较差, 可能是因为这些位点的甲基化程度值在各种体液中不存在1或0的绝对关系, 同时某些甲基化位点甲基化程度值会受环境影响而发生变化, 故根据不同样本筛选出的体液特异性位点会有差异, 需要通过实验对多个样本进行检测来验证其特异性是否足够稳定[20, 21]。在本研究中, 通过对其他3种体液共18个样本的鉴定结果, 证明所建体系的4个位点都具有良好的精液特异性, 只对精液DNA进行扩增。在位点的选择过程中, 引物设计同样具有重要的作用[22]。本研究所用引物包括精液特异性扩增与测序引物, 精液特异性扩增引物的选择需要综合考虑上下游引物上甲基化位点的分布个数及SNP的分布情况。同时由于转化过程对DNA的损耗, 目的片段长度不能过长, 否则可能会导致转化后的模板DNA片段扩增不出目的片段。本研究中有两个无法扩增出目的条带的位点, 其目的片段长度都超过了300 bp。测序引物的设计则需要尽量避免与相邻位点分型信息发生重合, 为此可进行正向或反向引物的选择。但是在选择反向引物后, 需要同时对电泳图上的分型信息进行转换, 如位点cg06045337。
确定了位点, 构建好体系后, 本研究对38个精液样本进行了鉴定, 结果证实该体系稳定性高, 对于各精液样本均做出了准确的鉴定, 呈现出完整的分型信息。然而由于样本数量的限制及其他民族样本的缺乏, 本研究中所取用样本在两位点cg06045337、cg06552563处未表现出多态性, 分别只出现了分型T和G。其与位点筛选时各SNP的MAF大于0.2的设定并不相符, 后续还应通过更多样本数据进行观察分析。此外, 该模型建立采用的样本为新鲜精液样本, 而实践中往往难以获得大量精液样本, 更多的为精斑, 因此需要评估该体系对于精斑的鉴定价值。本研究对6份于室温下存放15个月的精斑样本进行了检测。结果显示其中5份样本都出现了完整的分型, 表明该体系对于精斑样本仍有较强的适用性。但同时这也提示在使用该体系进行精液斑鉴定时, 若有部分位点未能呈现目标峰图, 需重新取样进行实验。虽然对于62份样本的鉴定结果显示非精液样本在目标位点处无峰图出现, 但对于部分位点无峰图的精液斑, 仍需更多样本数据进行辅助判断。同时在灵敏度测试中, 即使加入的转化后DNA量只有0.5 ng, 仍可检测得到完整的分型信息。当然, 还要注意到重亚硫酸盐转化的效率问题及在转化过程中模板DNA的损耗, 对于某些高度降解的样本, 该方法需谨慎选用。强行使用可能并不能得到完整的分型信息, 反而会导致珍贵样本的浪费。
本实验对不同体积比例的精液和阴道分泌液的混合斑也进行了鉴定。鉴定结果显示该体系对于法医实践中发现的混合斑中精液成分的检测有一定的价值, 其在精液与阴道分泌液的比例为1∶ 40时仍可达到较佳的检测效果。相较于Watanabe利用焦磷酸只能检测体积比例为1∶ 10的混合斑, 该法显然更具优势[18]。当然, 本研究所用混合斑检材为模拟检材, 如能使用在犯罪现场中搜集到的检材, 可更真实的反映该体系的应用价值。
综上, 本研究利用SNaPshot技术, 首次构建了以DNA甲基化为基础的精液特异性SNP分型复合体系。对62份样本的鉴定结果及对精斑、混合斑的实验结果, 显示该体系对于法医实践中精液鉴定有重要价值。当然, 该体系距离实用尚有距离, 仍有待完善:如引入管家基因对阴性结果判断是否存在生物检材; 进行种属确证实验, 验证是否具有人种属的特异性; 以及分析更多体液相关的甲基化位点数据, 补充更多的精液特异性位点, 使分型更加准确等。但同时也应看到基于DNA甲基化的复合SNP分型方法对体液鉴定的重大意义, 可以此进行其他体液特异性位点的探索, 从而构建更完整的以甲基化为基础的体液来源鉴定体系。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|