

{kind=link}
{kind=link}
{kind=link}
微流体液相色谱质谱联用分析头发中11种苯二氮卓类安眠镇静药物
[蒋文慧1  , 王朝虹
, 王朝虹2, * , 赵蒙2 , 李虹3 , 褚建新4 ]
, 王朝虹, 赵蒙|
|


第一作者简介:蒋文慧,女,浙江杭州人,学士,副高级任职资格,研究方向为法医毒物分析。E-mail: 836520133@qq.com
目的 建立新的微流体液相色谱质谱联用法分析头发中11种苯二氮卓类安眠镇静药物(劳拉西泮、氯硝西泮、溴西泮、氟硝西泮、阿普唑仑、替马西泮、氯氮卓、奥沙西泮、地西泮、7-胺基氟硝西泮和硝西泮)。方法 头发样品应用液液萃取法进行提取、微流控液相色谱质谱法进行检测。结果 11种苯二氮卓类安眠镇静药物的线性回归系数均大于0.99,分析精密度和提取回收率分别为2%~16%和71.7%~114.9%。最小检测限和最小定量限分别为0.008~0.03pg/mg和0.025~0.125pg/mg。结论 微流体液相色谱质谱联用法比传统的LC-MS/MS方法灵敏度高2~10倍。此方法已成功应用于实际毛发样本分析,可作为法医毒理学毛发中痕量成分检验方法。
Objective To establish a novel microfluidic LC-MS/MS method for analyzing 11 benzodiazepines (lorazepam, clonazepam, bromazepam, flunitrazepam, alprazolam, temazepam, chlordiazepam, oxazepam, diazepam, 7-aminoflunitrazepam and nitrazepam) in hair samples.Methods A simple liquid-liquid extraction (LLE) procedure was applied for preparing the hair sample, which was afterwards undergone into the LC-MS/MS analysis.Results A good linearity for the target analytes was obtained with the regression coefficients above 0.99. Precisions and extraction efficiencies were in the range of 2.0%~16.0% and 71.7%~114.9%, respectively. LODs and LOQs of the analytes ranged from 0.008 to 0.03 pg/mg and 0.025 to 0.125pg/mg, respectively.Conclusions The sensitivity of the microfluidic LC-MS/MS method is of ten-fold higher than the traditional LC-MS/MS methods, leaving a successful application to analyze a real hair sample, therefore able to be an alternative approach for forensic toxicological analysis of trace drug testing in hair.
Benzodiazepines are generally those prescribed medications, commonly used to treat symptoms of anxiety and insomnia[1]. These compounds have gained a wide clinic acceptance with outstanding effectiveness. However, the benzodiazepines would be significantly enhanced of their toxicity when taken in conjunction with either morphine, alcohol or any central depressants[2, 3], causing a series of accidents (e.g., the traffic) when misuse of them[4]. Due to their inherent adverse effects of high tolerance and dependence, benzodiazepines are severely abused by individuals or illegally used to commit criminal acts such as sexual assault[5, 6]. Therefore, detection of such drugs in biological samples might be very useful for the cases of drug-facilitated crimes. Generally, biofluids for drug testing include blood[7], urine[8] and saliva[9], all of which are routinely met in clinical and forensic laboratories. However, the biofluid concentration may only reflect the dosage at the sampling time. To assess compliance over longer periods or to trace back into medical history, it would be required to search for a non-invasive, easy-sampling and readily accessible specimen so as to provide a more permanent marker of drug intake.
Hair analysis has drawn much attention in recent years, together with its wide usage in various fields. With the appealing features over biofluids analysis, hair analysis has indeed become a complementary approach for the drug abuse testing[10, 11, 12, 13, 14, 15], pharmaceutical drug measurement[16], endogenous components analysis as well as environmental contaminants detection[17]. On one hand, collection of hair samples is non-invasive and free of embarrassment against the participants. Hair samples can be at room temperature to store without concern of chemical instability. On the other hand, hair sample is of a significantly prolonged time ready for detection since its harboring drugs and metabolites will remain for weeks or even months, contrasting to the drugs in biofluid samples only available for detection within several days after drug consumption. Therefore, hair analysis can not only provide more information about long-term assumption but also reveal the history of drug abuse. A number of analytical methods have been reported for identification and quantification of benzodiazepines in hair samples. Methodology includes gas chromatography-mass spectrometry (GC-MS)[1, 2, 4, 18], capillary electrophoresis-mass spectrometry (CE-MS)[19], high-performance liquid chromatography (HPLC)[20] and liquid chromatography-mass spectrometry (LC-MS)[3, 5, 15, 21, 22, 23]. Yet, their sensitivities are inadequate, with tedious and time-consuming derivatization steps required in some of these methods. Microflow LC was originally introduced in 1979[24]. Unfortunately, low-flow LC/MS has never been user-friendly and suffered from poor robustness. Since a single microchip, called ionKey which could incorporate with major LC components, was successfully designed and applied for the analysis of steroid hormones[25], pharmaceuticals[26] and peptide[27], the microfluidic device has reached a high sensitivity desired in a robust platform.
In this study, a microfluidic LC-MS/MS method was established for analysis of 11 benzodiazepines (lorazepam, clonazepam, bromazepam, flunitrazepam, alprazolam, temazepam, chlordiazepam, oxazepam, diazepam, 7-aminoflunitrazepam and nitrazepam) in hair samples. Compared with the traditional LC-MS/MS methods, the novel microfluidic LC-MS/MS has the advantages of less sample consumption, higher sensitivity and more environment-friendliness since 90% of solvent consumption can be saved for each sample. The developed method was successfully applied to analyze a real hair sample, too.
Standards of lorazepam, clonazepam, bromazepam, flunitrazepam, alprazolam, temazepam, chlordiazepam, oxazepam, diazepam, 7-aminoflunitrazepam, nitrazepam and diazepam-d5 (internal standard, IS) were purchased from Cerilliant (Round Rock, TX, US). Individual drug standards were prepared at 1 mg/mL in pure methanol, being stored at -20 ° C. All reagents were of HPLC grade. Acetonitrile and methanol were products from Fisher Scientific (Fairlawn, NJ, US). Formic acid was obtained from Spectrum Chemical MFG Corp. (Gardena, CA, US). Deionized water was generated with the Millipore Elix water purification systems (Millipore Corp., Molscheim, France).
Methanolic mixed standard solutions (1.0 μ g/mL) were prepared from each drug standards (1.0 mg/mL). The working standard solutions were prepared from the mixed standard solution through dilution to the appropriate concentrations as required. The working solutions were stored at -20 ° C.
Blank hair samples were obtained from healthy volunteers with no history of drug abuse. The donors have signed the informed consent. Hair samples were stored in a clean envelope at room temperature. All the procedure regarding with the sample collection was approved by the Ethics Committee on Human Research of the Faculty of Health Science.
Prior to analysis, the hair samples were firstly washed with shampoo and secondly rinsed with deionized water then acetone and finally air-dried overnight. The washing solutions were analyzed via the novel microfluidic LC-MS/MS methods to secure no presence of contamination. Dried hair sample was cut into 1 mm pieces and ground with a ball-grinder (Retsch, Haan, Germany). 20 mg of ground hair sample was weighed in a glass tube, with addition of 100 μ L of mixed standard solution and 2 mL of boric acid buffer (pH 9.5). The mixture was ultrasonicated for 30 minutes before addition of 2 mL of extractant (dichloromethane/ether/hexane=30/50/20), being undergone with vertex for 10 minute. The extract was centrifuged at 10 000 r/min for 5 minutes. The supernatant was transferred to a clean Eppendorf tube and was evaporated under N2 stream until almost dry. The residue was reconstituted by 50 μ L of methanol/water (1꞉9), with 1.0 μ L for the analysis by either the novel microfluidic LC-MS/MS or the traditional LC-MS/MS system.
The analysis of 11 target drugs was performed with Waters ACQUITY UPLC M-class combined into the Xevo TQ-S mass spectrometer that was fitted with an IonKey source. The analytes were separated on an iKey peptide BEH C18 column (150 μ m× 50 mm, 1.7 μ m; Waters, US), which was held at 45 ° C. Mobile phase A was water with 0.1% formic acid. Mobile phase B was consisted of methanol (containing 0.1% formic acid) and acetonitrile (3꞉1). The mobile phase was changing from the initial of 10% B to the final 95% B in 6 min and kept for 2 min, followed by a descending linear gradient to 10% B in 0.5 minutes and held constant for 4.5 minutes. The flow rate was 3 μ L/min. The UPLC M-class system was connected to Xevo TQ-S MS equipped with an IonKey source in positive mode. The MS parameters were listed as: capillary voltage 3.8 kV, the source temperature 150 ° C and the cone gas flow 50 L/h. 1 μ L of the hair extract was subjected into the novel microfluidic LC-MS/MS system and the chromatographic data were recorded by MasslynxTM software (Version 4.1).
The analysis was performed by Waters ACQUITY UPLCTM coupled with Xevo TQ-S mass spectrometer (ESI source). The 11 target drugs were separated through a BEH C18 column (2.1μ m× 50 mm, 1.7 μ m; Waters, USA), which was maintained at 45 ° C. Mobile phase A and B were the same as those mentioned in section 2.5. The gradient was slightly adjusted. The initial mobile phase composition (2% B) was kept for 0.5 minutes and increased to the 95% B in 5.5 minutes with another 2 minutes kept, followed by a descending gradient to 2% B in 7 minutes and held constant for 2 minutes. The flow rate was 0.5 mL/min. The UPLC system was connected to the Xevo TQ-S MS equipped with ESI source in positive mode. The MS parameters were the following: capillary voltage 3.0 kV, the source temperature 150 ° C , the desolvation temperature 500 ° C and the desolvation gas flow 800 L/h. 1 μ L of the hair extract was undergone into the traditional LC-MS/MS system and the chromatographic data were recorded by MasslynxTM software (Version 4.1). The two transitions per analyte and respective setting for LC-MS/MS system were given in Table 1.
| Table 1 MRM transitions and respective settings for each analyte |
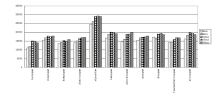
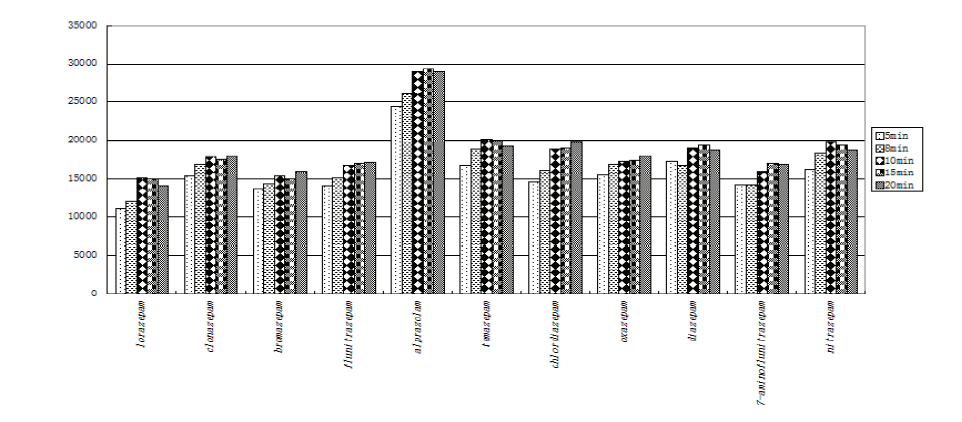
There are several factors affecting the extraction efficiency, e.g., ultrasonic time, extraction solvent and extraction duration. The influence of these factors was studied and optimized. A KQ-250E ultrasonic apparatus (Kunshan, China) was used for isolation of the 11 benzodiazepines from hair sample. 100 μ L of mixed standard solution was spiked into 20 mg of ground hair. Then, 2 mL of boric acid buffer was added as reported from literature [19] for reference. The ultrasonic process was carried out at 50 W for 10, 20, 30, 40 and 50 minutes, respectively. It was showed that extraction efficiency achieved best at 30 minutes (shown in Fig.1).
 | Fig.1 The effect of ultrasonic time on extraction efficiency |
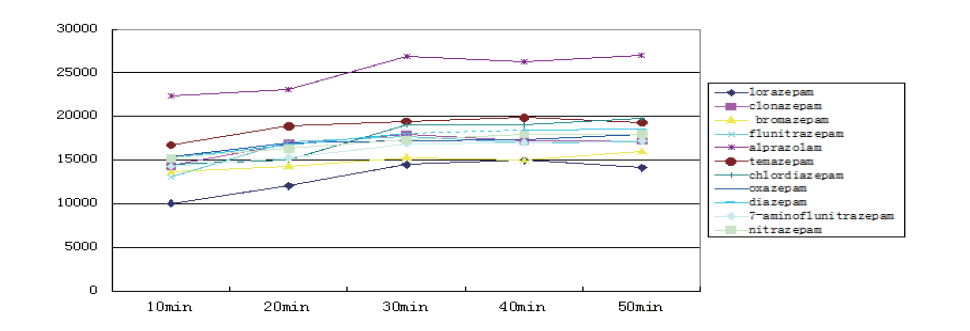
The liquid-liquid extraction process was another important factor for the extraction efficiency. Several solvents such as ethyl ester, dichloromethane, ether and hexane were examined. According to the results, a mixture of dichloromethane/ether/hexane (30/50/20) was found to get the highest extraction efficiency and thus selected as the extractant. The influence of liquid-liquid extraction time was also optimized in the range of 5~20 min, demonstrating that the extraction efficiency remained constant after 10 min. Therefore, 10 min was chosen for the following work (shown in Fig.2).
 | Fig.2 The effect of extraction solvent/time on extraction efficiency |
According to FDA guidelines, both the novel microfluidic LC-MS/MS and the traditional LC-MS/MS were validated on the items of their selectivity, specificity, linearity, limit of quantification (LOQ), limit of detection (LOD), precision, accuracy, extraction efficiency and matrix effect. Selectivity and specificity were evaluated with analysis of six different sources of blank hair samples and spiked hair samples at the concentration of 10 pg/mg through the novel microfluidic LC-MS/MS method and the traditional LC-MS/MS choice, respectively. Mean area measurements for the selected analyte ions were generated and analyzed to ensure that the matrices were free from interfering compounds. Calibration curves were prepared by the spiking of appropriate mixed standard solution into 20 mg of blank hair at concentrations of 0.005, 0.012 5, 0.025, 0.05, 0.125, 0.25, 0.5, 1.0, 2.5, 5.0 and 10 pg/mg for each analyte. Calibration was performed through linear regression of peak-area ratio of the targeted drugs and IS versus the respective standard concentration. LOQs corresponding to the lowest concentrations were used for the calibration curves under a signal-to-noise ratio of at least 10. The resulting linearity ranges, correlation coefficients, LOQs and LODs of each analyte were reported in Supplementary Table S1. A representative chromatogram of LODs for each compound was showed in Fig.3.
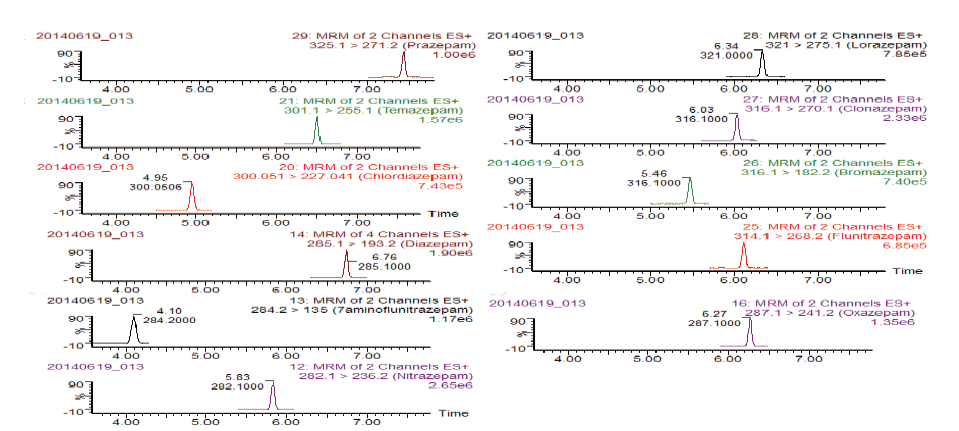 | Fig.3 Quantitative ion chromatograms of extracted hair samples harboring the 11 drugs at LOQ |
The precision and accuracy were evaluated at three concentration levels: lowest, middle and highest points of the calibration curve from standards. The intra- and inter-day precisions were evaluated via analysis of five replicates of each level for three consecutive days. The accuracy was the percent of calculated concentration value at a specific level from the respective nominal concentration. Data of precision and accuracy were listed in Supplementary Table S2.
Ion suppression was estimated for each analyte through comparison of the peak area obtained from hair samples spiked before extraction, against that of a methanolic standard at the responding concentration. The extraction efficiency was determined for each analyte through comparison of the peak area obtained from hair samples spiked before extraction, against that obtained from hair samples spiked after extraction. The data were showed in Supplementary Table S3.
Both the novel microfludic LC-MS/MS method and the traditional LC-MS/MS have a good linear relationship for each analyte, with their correlation coefficients all above 0.99. For the novel microfluidic LC-MS/MS, it was showed of a wide linear range. All of the analytes displayed 2~10 folds higher of LODs from the novel microfluidic LC-MS/MS method than the traditional LC-MS/MS one. The intra- and inter-day precisions via the two LC-MS/MS methods were comparable and within the required limits of 15% RSD. Accuracy ranged from 88.4% to 113.6% for the novel microfluidic LC-MS/MS method and from 93.6% to 110.9% for the traditional LC-MS/MS, respectively. Supplementary Table S4 showed that the extraction efficiency and matrix effect were also satisfactory.
Early in 1997, V. Cirimele et al[2] presented a GC-MS method for the detection of human hair of forensically relevant benzodiazepines. The LODs only reached 1~20 pg/mg. A. Salomone et al[5] developed and validated a specific LC-MS/MS protocol for determining benzodiazepines at low concentration in hair specimens. For diazepam and bromazepam, the sensitivity was improved significantly. C. Montesano et al[16] established a UPLC-MS/MS assay for analysis of 96 drugs including benzodiazepines and the LODs ranging from 1 to 20 pg/mg. Recently, A. Woź niakiewicz et al[22] developed a quick CE-MS/MS method for simultaneous determination of benzodiazepines in hair samples. The LODs for benzodiazepines were within 6~23 pg/mg. Obviously, the sensitivity of reported methods could not fit for our purpose about trace amount of benzodiazepines detection. Both clear and intuitive comparison data were summarized in Supplementary Table S5. An effective increase was observed in sensitivity with the developed microfluidic LC-MS/MS method as compared to published literatures. With the ultrasensitive microfluidic LC-MS/MS method, more information about long-term assumption would be provided through hair analysis.
The sensitivity improvement of the microfluidic LC-MS/MS method is attributed to the novel ionKey source. Firstly, an electrospray plume generated from conventional LC flow rates can be quite broad and divergent. As the solvent is reduced of its flow rate, the electrospray plume decreases in size and becomes more convergent. This allows the inlet of the mass spectrometer to become more efficient and capture a greater percentage of the plume, resulting in the increase of ion signal. Secondly, smaller charged droplets are produced by ionKey source so that ions are much more preferential to gather on the surface of charged droplets and are therefore further more prone to be gasified. This improves the ionization efficiency. Thirdly, the decreasing droplet size causes fewer interfering charged ions when ionization in each droplet and thus ion suppression is reduced. Based on the three points mentioned above, a dramatic sensitivity improvement has been successfully achieved by the microfluidic LC-MS/MS method.
A hair sample was collected from a 45-year-old woman who had taken 1mg of Flunitrazepam every night to treat insomnia for 2 weeks. She has no other medical history. The hair sample was cut close to the scalp from the posterior vertex area of the woman’ s head, and prepared according to the above-mentioned protocol in section 2.4. Subsequently, the extract was submitted for analysis. Flunitrazepam and its metabolite 7-aminoflunitrazepam were detected, with their concentrations being respective of 1.5 and 4.2 pg/mg, such low detection limits that the traditional methods are usually unable to reach.
In this study, 11 benzodiazepines in hair samples were quantitatively analyzed with the microfluidic LC-MS/MS method. The established method was obtained of comprehensive and satisfactory results. Compared with the published literatures, a dramatic increase was achieved in sensitivity. The proposed method was successfully applied into a real hair sample for drug screening, demonstrating the utility of the newly-established microfluidic LC-MS/MS method as well as providing an alternative strategy for testing trace drug in hair.
For the supplementary materials of this article, please see: http://www.xsjs-cifs.com/CN/volumn/home.shtml.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|