

{kind=link}
{kind=link}
{kind=link}
{kind=link}
EA-YPredictor:基于Y-STR数据的家系特异性单倍群归属判别分析软件
[殷才湧1, 2, 3  , 孙辉
, 孙辉4 , 周怀谷5 , 金力1, 2, 3, 6 , 李士林1, 2, 3, 6, * ]
, 孙辉]
|
|


第一作者简介:殷才湧,男,江苏泰州人,博士研究生,研究方向为Y染色体遗传标记法医学应用。E-mail:cyyin18@fudan.edu.cn
目的 Y染色体为男性所特有,其遗传标记蕴含着丰富的生物地理信息,故可溯源家系,在嫌疑人排查和追踪中发挥作用。Y-STR突变率较高,而Y-SNP突变率极低,几乎不会发生回复突变,所以后代男性群体携带祖先特有的Y-SNP。本研究期望通过现在我国Y库建设中通用的17个Y-STR的单倍型数据预测Y-SNP单倍群细支。方法 基于前期观察,选取千人基因组计划III期中的513例东亚人群(中国及周边区域)作为基础数据集,在Java平台和Microsoft Excel软件框架下,以遗传距离计算和Y染色体进化树构建手段相联合研发Y-STR数据的家系特异性单倍群归属判别分析软件:EA-YPredictor。结果 本研究揭示了15个单倍群大支下的核心单倍型。通过随机选取70个公开数据库样本,EA-YPredictor软件预测准确性达到92.8%(95%置信区间:[84.1%, 97.6%])。结论 在Y-SNP复合扩增检测尚无定论的情况下,本软件可基于二代测序样本对Y-STR数据库样本进行单倍群细支的准确预测,能适用于辅助家系单倍群判断。随着测序技术的不断换代和优化,更多高通量的Y-STR和Y-SNP数据补充将会使本软件进一步优化。此外,本软件对于Y数据库中Y-SNP遗传标记的筛查建库有一定指向作用。
Y chromosome, male specific, carres the genetic markers that could indicate the bio-geographic information of unknown male individuals, therefore playing vital and unique roles in pedigree searching and individuals tracking. Y-SNPs, the genetic markers hardly occurring of reverse mutation, harbor extreme low mutation rate compared to Y-STRs. Thus, male individuals of the same lineages always carry the specific Y-SNPs of their identical ancestors. However, Y-SNP typing is time- and cost- consuming, making it not an ideal tool for investigation. To target suspects’ pedigree for investigation purpose, Y-SNP haplogroups were here tentatively predicted from the current 17 Y-STR haplotypes that are commonly used for Y-database construction. With re-evaluation of previous research results and analysis of the open East-Asian datasets from 1000-Genome Project (Phase III), one new Y-STR-based pedigree-specific haplogroup prediction software, EA-YPredictor, was developed through calculation of genetic distance and Y chromosomal phylogenetic tree reconstruction, hence successfully having screened the 15 Y-SNP major haplogroups out from the core 17 Y-STR haplotypes on the adaptable Java platform or Microsoft Excel formulation. Randomly selected of 70 new samples, EA-YPredictor was tested of its prediction accuracy (92.8%, 95% CI: [84.1%, 97.6%]), revealing the exact Y haplogroup affiliation to the males of East Asian ancestry, thereby demonstrating its validity to predict the Y-SNP haplogroup of samples in existing Y chromosome database. Following the next-generation sequencing technology to confirm the location and annotation of more Y-STRs and Y-SNPs, EA-YPredictor will be definitely optimized further so that more indications would be provided on screening which set of Y-SNP markers from Y chromosome databases.
快速准确地推测男性犯罪嫌疑人的家系和来源群体, Y染色体数据库(Y Chromosome Database, 简称Y库)能发挥重要作用, 这由其雄性(Holandric)或Y连锁遗传(Y-linked Inheritance)[1]的特征所决定。Y染色体95%不能重组交换的区域即非重组区(Non-recombining Region, NRY), 在不考虑突变的情况下, 其变异能够完整地遗传给男性子代[2], 其中为物证鉴定研究最多的是Y染色体短串联重复序列(Y Chromosome Short Tandem Repeat, Y-STR)和Y染色体单核苷酸多态性(Y Chromosome Single Nucleotide Polymorphism, Y-SNP)。
Y-STR是现阶段Y数据库所采用的遗传标记。Y-STR单倍型(haplotype), 能用于男性家系判别。由于不同的Y-STR基因座突变速率不同, 不同Y-STR基因座组合产生的单倍型和家系辨别能力会有差异[3]。实际工作中, 最早应用的试剂盒是AmpF/STR® Yfiler® 试剂盒(美国Thermo Fisher公司), 能一次复合扩增17个STR基因座, 包含了欧洲小单倍型(Minimal Haplptype)的核心基因座, 即由DNA分析方法科学工作组(SWGDAM)所推荐的基因座和6个高突变率基因座。近年, 多个商业化试剂盒的Y-STR复合检测体系也均包括上述17个基因座, 如PowerPlex Y23(23个Y-STR基因座, Promega公司)和AmpFlSTR® Yfiler® Plus Kit(27个Y-STR基因座, Thermo Fisher公司)。在全国范围内排查最通用的Y-STR单倍型时, 上述17个STR基因座是最好的选择。通常情况下, 现场嫌疑人的所有父系亲属(兄弟, 父亲, 儿子和叔伯等)与其Y-STR单倍型应是一致的, 然而, 在复杂男性家系及远房亲属之间, 却存在Y-STR单倍型在个别高突变基因座上的差异。此外, 即使非同源男性的Y-STR单倍型, 也可能在高突变基因座上差异比较少。这些实际问题是家系排查工作中遇到的挑战。Y-SNP具有极低的突变速率(约为2× 10-8)[4], 以至于几乎不可能出现回复突变。从理论上来说, 现代人类男性拥有最近的共同祖先“ Y染色体亚当” [5], 因而成为现代人类所有Y染色体的来源。在父系社会中, 除去收养、过继和非婚生等特殊情况, 理论上同一男性祖先的男性后代具有相同的Y-SNP单倍型, 这为以Y-SNP遗传标记排查男性父系谱系奠定了基础。Y-STR单倍型相近、携带同样的Y-SNP突变体的样本, 构成Y染色体单倍群(Y Chromosomal Halpogroup)。Y-SNP单倍群随着人群的迁徙和繁衍而不断扩散, 最终形成地域的普遍性, 而扩散的过程中也不断出现新的Y-SNP突变, 这些突变大多数依照一定的时间顺序发生, 最终成为记录人类进化史的分子钟。由此看来, Y-SNP在男性家系辨别能力方面要优于Y-STR。但是, 开发适用于物证鉴定的Y-SNP复合扩增体系尚待时日。本课题组提出在东亚范围内, 对我国现有Y库样本单倍型数据进行Y-SNP单倍群细支预测, 在此基础上通过实验手段验证预测结果, 以期为犯罪现场嫌疑人或受害者的家系排查提供可依据的推断结果。
通过Y-STR数据推测Y-SNP单倍群归属是具前景价值的, 2005和2006年, Athey首次通过遗传距离和贝叶斯等位基因频率的方法分别发布了Y单倍群预测软件, 这也是公认准确率较高的现有软件[6, 7]; 亚利桑那大学的单倍群划分软件则采用了机器学习方法[8]。但上述方法所采用的数据集主体是西北欧、东欧、地中海、南亚和美洲人群。考虑到以东亚人群为遗传背景、基于Y-STR数据准确推断Y-SNP的软件在生物地理来源推测中应会发挥作用, 本实验室自1998年开始联合运用Y-SNP和Y-STR遗传标记推断东亚人群的迁移路线和历史事件发生时间, 如首次揭示东亚人群的非洲起源[9]。经过多年积累, 我们针对中国人群筛选出了434个Y-SNP单倍群分支, 可以覆盖所有东亚男性家系。C、D、N和O单倍群大支下的分支数目分别为130、14、43和203。此外, 在不同单倍群分支上, 男性个体数目有很大差异。因而, 经过若干代传递后, 这些Y染色体单倍群内的Y-STR单倍型有显著差异。为了提高这些单倍群分支所属上游单倍群的准确率, 我们还在单倍群人数占比较高的分支添加了79例个体。后续研究中, 随着更多高通量数据的补充, 我们也会在其他分支上进一步增加个体数。以上这些分支是通过Y染色体全长测序数据进行命名的, 已上传到ISOGG的Y染色体进化树(2018年, 版本号:13.01)。基于充分确认的Y-STR单倍型与Y-SNP单倍群的连锁状态, 本研究以17个Y-STR单倍型对未知来源的男性个体的Y-SNP单倍群进行推断, 希冀有助于排查男性嫌疑人家系、缩小排查范围。本文选取了二代测序深度和准确度都相对较高的千人基因组计划III期[10, 11]中的513例样本, 这些样本的Y-SNP单倍群在系统发生树上的位置十分深入, 适合作为本研究中Y-STR预测Y-SNP数据的基础。该软件具备开创性, 提出了一个全新的建立Y染色体数据库的思路, 可同时涵盖Y-SNP和Y-STR两种遗传标记, 并且允许数据库本身对原始数据进行加工整合, 以得到新的信息。本程序完全开源和开放, 允许对数据库进行各种类型的增补修改, 原始Y-SNP和Y-STR数据以及Y-SNP进化树也允许随时更新。尽管软件的设计目的是对家系进行鉴别, 但它的衍生程序也能用于Y-STR核心单倍型的计算和Y-SNP单倍群的建树。随着二代测序技术的不断换代和优化, 以及大数据时代的到来, 在未来有更多Y-STR基因座和更精确Y-SNP定位的前提下, 补充更多有价值的参考数据, Y-SNP单倍群的鉴别将会更加精确。
前期本实验室对部分进行了Y染色体全长测序的样本和千人基因组计划(1000 Genomes Project phase III)的Y染色体数据进行观察, 充分确认了Y-STR的单倍型与Y-SNP单倍群的连锁状态。数据库原始数据来源于千人基因组计划III期中搜索并分析的东亚区域Y-SNP和对应Y-STR数据, 综合根据Y单倍群分支、地理定位(东亚及周边区域)和Y染色体序列信息筛选了513例遗传多样性和地理代表性较强的样本(来自434个Y-SNP单倍群细支, 部分分支包含多个个体), 以求拓宽中国男性个体Y-SNP单倍群预测的范围并提高其可信度, 数据库总量为513例个体。本研究所采用的基础和测试数据都来源于Y染色体的海量全测序列, 因而虽然个别Y-SNP分支样本量较少, 但是其应具有高准确性和可重复性。
源数据中Y-SNP单倍群的分类十分精确, 各单倍群样本量过少, 因此统一以单倍大群下第二级亚群的祖先型分类, 用于计算对应于每个Y-SNP单倍群分类的Y-STR核心单倍型。二级亚群内某个与其他单倍型遗传距离最近的单倍型即为核心单倍型。源数据中Y-STR单倍型仅统计17个Yfiler试剂盒基因座。
Y-SNP单倍群可以构建系统发生树, 它对应的或位于它下游的Y-STR单倍型可以追溯到Y-SNP单倍群的枝上。通过计算未知的Y-STR单倍型与数据集中其他Y-STR之间的遗传距离, 就能够判断其在系统发生树上的位置。两个Y-STR单倍型遗传距离d的计算公式[12]如下:
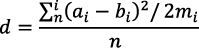
其中n为参与计算的Y-STR基因座数目, ai为待测样本A第i个Y-STR基因座的数值, bi为已知样本B第i个Y-STR基因座的数值, mi为第i个Y-STR基因座的突变速率, 本文所涉及的17个Y-STR突变率综合参考了三个中国汉族突变率的研究结论[13, 14, 15]。需指出, 待测样本A和已知样本B的Y-STR单倍型其参与计算的Y-STR基因座必须是相同的。
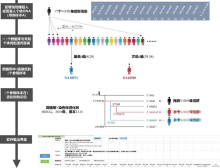
将推断的步骤和算法以流程图展示(图1)。已知待测样本A和数据库中每一个已知样本之间的遗传距离, 则通过对遗传距离排序, 可明确与A最接近的两个已知Y-SNP单倍群归属的样本, 从而可根据预设的ISOGG公布的Y-SNP树(版本号:13.01, 2018年)推断待测样本A可能的Y-SNP单倍群。
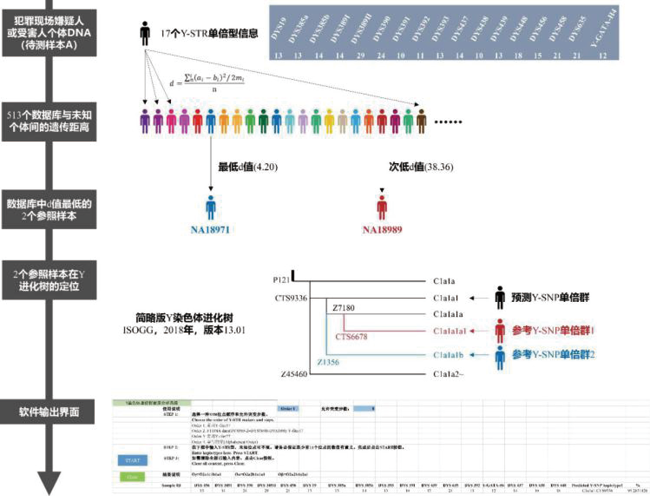 | 图1 Y-SNP单倍群预测模型和算法Fig.1 The Y-SNP haplogroup prediction model and algorithm underlying this software |
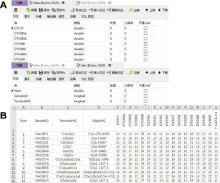
由于本数据库的规模有限, 且不需要在数据层借助存储过程或使用服务器满足计算需求, 本着易于移植和二次开发的目的, 采用相对精巧的MySQL数据库作为储存和读取数据的基础。建立数据库CDNO, 为方便下游程序, 数据库分为Data和Info两个数据表。其中Data表格负责储存Y-STR单倍型数据, 栏位名为Y-STR基因座名称, 数据类型为双精度浮点型(double)。Info表格负责储存Y-SNP单倍群数据, 包含主键, 栏位名包括样本编号(Sample ID)、最终单倍群(TerminalHG)和主单倍群(MajorHG), 数据类型均为长文本型(longtext)。图2A是Data表格和Info表格对变量进行定义和限定的界面。图2B是数据库中的千人基因组计划实际样本的Y-SNP单倍群和Y-STR单倍型信息, 每行为一例样本。
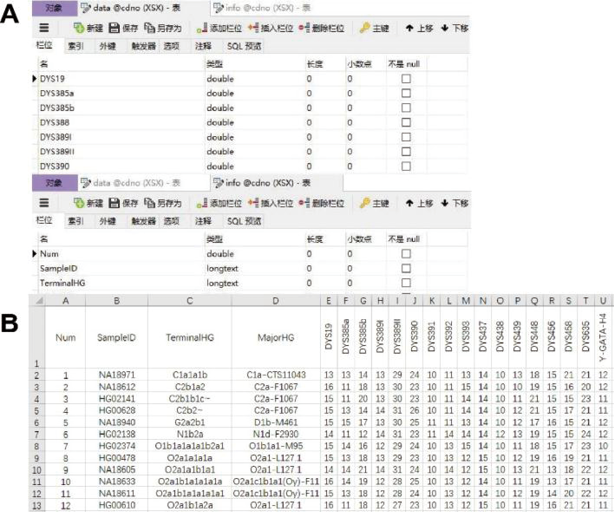 | 图2 MySQL数据表格格式Fig.2 MySQL-formatting representation of 17 Y-STR loci and the correlating Y-SNP haplogroups |
数据库读取和计算的实现依托于Java的MySQL connector模块。先从MySQL数据库中读取全部Data表格作为二维数组, Info表格作为对象列联表, 再通过对数组进行数学计算, 结果返回MySQL数据库中查询对象。软件定名为EA-YPredictor, 开发基于Eclipse。
由于Java程序对环境十分依赖, 使用较不方便, 为了更好地推广使用, 将其移植到Excel环境中, 并增补一些适用于Excel的功能。Excel移植版软件命名为EA-YPredictor.xlsm。
1.6.1 数据库模块
Excel本身就具有数据存储的功能, 数据表分为两部分。工作表DATA存储样本Y-SNP单倍群和Y-STR单倍型数据以及Y-STR基因座突变速率数据。工作表SNP则存储了最新的Y-SNP单倍群树。
1.6.2 功能实现模块
功能模块主要依赖工作表Count和Result。Count表格内置公式, 用来计算待测Y-STR单倍型和参考样本Y-STR单倍型之间的遗传距离, VBA程序将排序后的Y-SNP单倍群结果输出到工作表Result中。工作表Matrix将抽象的Y-SNP树转化为直观的二进制矩阵, 通过VBA程序定位结果单倍群位置, 并输出到使用界面中。
1.6.3 使用界面部分
使用界面主要是用户界面工作表User’ s, 这是程序中用户唯一可见的输入界面(图3)。
按照说明使用, 输入待测Y-STR单倍型, 输出结果如图4所示。
 | 图3 Excel版用户界面示意图Fig.3 Diagram of user’ s interface in Excel version |
 | 图4 Excel版结果输出示意图Fig.4 Diagram of resultant output in Excel version |
1.6.4 功能更新优化
相比于早期的Java版本, Excel版本增补了一些功能, 并进行了算法的优化。在这个版本中增加了定位祖先Y-SNP单倍群和标记差异性两个功能, 并针对参考集和测试集二代测序数据中DYS385a/b转座的现象进行了优化:将待测和参考样本的DYS385a/b基因座上的两个等位基因进行判断, 即根据等位基因长度排序后再计算遗传距离, 从而避免DYS385a/b基因座转座引起的遗传距离高于真实水平的情况。现在可以将和待测Y-STR单倍型最近的两个参考单倍型的Y-SNP单倍群结果向上追溯到共有的一个最近祖先型Y-SNP上, 从而大大提高了定位结果的准确性。当两个参考的Y-SNP相距过远时, 会自动标记为星号(* ), 并忽略DYS385a/b基因座的结果重新进行计算, 避免因为DYS385a/b基因座出现转座的情况导致Y-SNP定位结果出现较大误差。
Y-STR核心单倍型是与所有Y-STR单倍型遗传距离最小的一种Y-STR单倍型。计算基于Excel VBA程序, 结果见表1。
为了评价该软件的准确度, 本研究选取了70份新的千人基因组计划东亚人群数据验证单倍群细支预测的准确性(数据尚未发表, 故不全部列出)。其中准确判断的有65份, 未能准确判断的5份原始数据见表2。对于70份东亚人群数据的检测结果显示的单倍群细支预测准确度为92.8%(95%置信区间:[84.1%, 97.6%]), 适用于辅助家系单倍群判断。从表中可见, 样本GH320实际单倍群为J1, 由于参考数据库中没有任何J类单倍群数据, 故无法准确判断。样本GH323和HZJ1980虽然能够定位到单倍大群甚至亚群, 但由于该样本是罕见的独特单倍群, 在参考数据库中没有近似, 也未能准确判断。而样本GH348和HZJ1981都属于单倍群R1b1b1, 其祖先型单倍群R在参考数据库中数据量很少, 所以被错误归类到单倍群O2a中。
| 表1 Y-SNP单倍群大类对应的Y-STR核心单倍型 Table 1 The major Y-SNP haplogroups and their correlating core Y-STR haplotypes |
| 表2 5例预测错误样本的预测及实际检验结果 Table 2 Comparison of the predicted and actual (by testing) Y-SNP haplogroups from 5 falsely predicted cases |
从遗传学而论, Y-STR的突变速率更快且存在回复突变, 而Y-SNP相对稳定, 故而使得Y-SNP单倍群下对应着不止一种Y-STR单倍型。如果将这个家系视为一棵树, Y-SNP单倍群就是树干, Y-STR单倍型就是树枝, Y-SNP处于Y-STR的上游。经过精确测量的Y-SNP单倍群可以依据SNP位点的突变时间构建出系统发生树, 它对应的或位于其下游的Y-STR单倍型可以追溯到Y-SNP单倍群的枝上。当有一个新的Y-STR单倍型出现时, 通过计算它与其他Y-STR之间的遗传距离, 就能够知道它在系统发生树上的位置。而Y-SNP单倍群的“ 枝” 越深入, 也就表明Y-SNP突变出现得越晚, 其单倍群中的Y-STR单倍型出现的变异型就会越少, Y-STR和Y-SNP的相关性也就越强, 甚至可以达到一一对应。
在法医学实践中, 虽然Y-SNP的分型工作普及尚低, 但本研究开发的通过Y-STR单倍型预测Y-SNP单倍群的软件, 对东亚人群的判定准确率可达92.8%, 可为案件侦破提供思路和新借鉴。513个来自东亚人群的男性个体均有Y-STR和Y-SNP测序数据, 精确地显示出了每个个体的Y-STR和Y-SNP基因型, 这些样本所处的Y-SNP单倍群属于进化树内中国人群主要分布的大支, 具有显著的群体代表性。此外, 根据不同分支所代表群体的数量不同, 本研究在群体数量比较大的单倍群分支中纳入了较多详细的子分支, 充分考虑了东亚人群Y-SNP单倍群的特征。通过对Y库样本进行大样本的预测, 本软件对于我国Y-SNP遗传标记的筛查建库应能提供方向导引。
本软件在设计之初便考虑了民族多样性, 因而选择了东亚及周边区域样本。各民族的历史形成及当代多民族人群融合结构虽然不尽相同, 但值得注意的是, Y-SNP单倍群进化树是基于Y染色体遗传过程中SNP位点累加突变形成的, 从主干的分支到末端可能存在5~10个分支(如O2→ O2a2b1a2a1a)。人群的Y染色体单倍群分支可能经过若干代后产生若干个细支, 故可根据突变的Y-SNP位点划分细支。因而, 本软件推断的结果是家系特征性Y-SNP, 与样本来源的民族关系较小。
但不可忽视的是, 该软件目前仍存在问题。首先, 受限于Y-SNP单倍群测序的难度, 软件基底数据量较少, 数据库目前C、D、N、O四个单倍大群的数据相对更多, 其他一些东亚人群中不太常见但欧美和非洲人群中很普遍的单倍大群如E、F、H、J、R、Q等的样本很少或就没有, 分型也非常浅。由于近代世界范围内发生了数次人口迁移, 不仅在经济文化在基因层面也发生了东西方的碰撞交融, 仅用几个东亚常见的单倍大群去预测全部东亚人群存在着很大的不准确因素, 所以该软件应主要针对或用于中国男性人群。其次, 随着对Y-SNP单倍群研究的深入, 越来越多的单倍群支系被发现, 仅用数据库中位于它们上游或平行处的单倍群是无法准确预测新型单倍群的, 这些困难会在实际检案中逐渐浮现并得到解决。
现阶段, Y-STR数据库中纳入的Y-STR基因座取决于不同用途, 根据Kayser研究团队的成果[4, 16], 低突变率Y-STR基因座主要在家系的筛选中发挥作用, 而高突变率基因座则被更多地应用于识别家系内部不同的男性个体。Y-STR基因座两两之间都处在连锁不平衡状态, 但是具体的连锁机制及其在人群中的变化情况仍待探索。本软件关注的是Y-STR组合推断Y-SNP单倍群的可实现性和准确性。据不完全统计, 国内的建库方案几乎都是建立在这17个Y-STR基因座的基础之上, 本研究为了同时考虑Y-STR基因座与Y-SNP单倍群的相关性以及位点的推广程度, 采用这17个Y-STR基因座开发了该预测模型。鉴于Y-STR的突变率较高, 后续的开发工作需要重点评估参考集人群的数量和多样性对Y-SNP预测的准确性影响, 动态观察参考集人数与准确性的关系, 以进一步提高系统的可靠性和效能。
此外, 本软件采纳的Neis遗传距离模型[12]是基于无限等位基因模型(Infinite Allele Model, IAM)[17]。而具有相同Y-SNP单倍群归属的家系中, Y-STR会随机发生Y染色体微变异、拷贝数变异和等位基因缺失等小概率变异。如日本研究团队的结果[18]显示:DYS458-14.1/16.1等位基因均来源于N单倍群, 另也有一部分DYS458-14等位基因的个体属于N单倍群。DYS458-14和15比DYS458-14.1在人群中具有更高的频率, 说明与微变异相比, 单个基序突变的可能性更高, 而14.1微突变具有很明显的家系特异性。在非洲人群[19]DYS385基因座出现的13.2和14.2微变异等位基因, 两者等位基因频率之和为6%; 相比之下, 我们在江苏宿迁人群(n=8592)中发现了DYS385-13.2的等位基因, 因此, DYS385-13.2不具有群体特异性, 而具有家系特异性。综上, 对于一些复杂家系水平发生的变异, 应考虑其家系特异性。
在应用方面, 图1主要展示本软件的应用层面和流程图。本软件基于17个Y-STR基因座预测的Y-SNP单倍群结果包括3种可能性较高的单倍群分支, 刑事案件中的犯罪嫌疑人或受害者的Y-STR数据, 经本软件推测的单倍群分支可能来自于3种不同的分支。对于不同分支, 需筛选更加详细的下游分支并采用方便快捷的Y-SNP分型手段进行验证。在未来软件开发中, 本研究团队会根据实际需求, 提高应用场景中的数据运算量并优化软件环境和数据库效率等以利于进行大规模的运算。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|