
{kind=link}
固相萃取结合超高效液相色谱质谱方法同时测定尿中的6种苯丙胺类毒品
[王朝虹1  , 张琳
, 张琳1 , 赵蒙1 , 李虹2 , 刘帅3 ]
, 张琳|
|


本文建立了固相萃取-超高效液相色谱-质谱法(SPE-UPLC-MS/MS)同时测定尿中6种苯丙胺类毒品。尿样经过Oasis MCX 固相萃取柱提取净化,应用电喷雾离子源(ESI)、多反应监测(MRM)正离子模式质谱进行定性及定量检验。6种苯丙胺类毒品在0.1~20 ng/mL范围内线性关系良好,相关系数优于0.9987,提取回收率大于78 %,RSD小于10 %。本方法简单、快速、灵敏、准确、可应用于尿液中痕量苯丙胺类毒品的分析。
Author: Wang Zhaohong (1974—), female, doctor, professor, mainly focusing on research and detection of trace evidence. E-mail:wangzhaohong27@163.com
A rapid and sensitive method has been developed for simultaneous determination of 6 amphetamine-type drugs in urine sample by solid phase extraction-ultra performance liquid chromatography coupled with mass spectrometry (SPE-UPLC-MS/MS). The samples were extracted with Oasis MCX solid phase extraction cartridge and analyzed with the UPLC-MS/MS system coupled with an electrospray ionization (ESI) source, finally having been measured in ESI+ mode and multiple reaction monitoring (MRM). Six amphetamine-type drugs were linear in the range of 0.1~20 ng/mL, with correlation coefficients over 0.9987. The recoveries were above 78 % with RSD less than 10.0%. The method is simple, rapid, sensitive and accurate for the determination of trace amphetamine-type drugs in urine sample.
Amphetamines, a class of synthetic organic amine compounds, are powerful central nervous system stimulants [1]. Abuse of amphetamines shows an increasing trend in the world and has become a global problem in recent years [2]. These drugs are usually prescribed for illicit drugs and are strictly prohibited in China since they severely damage human health in both mental and somatic aspects [3, 4]. However, their recreational use is still prevalent, mainly among youth, despite warnings of irreversible damage to the central nervous system [5].
Amphetamines’ test is routinely performed in clinical and forensic laboratories by screening the biological samples including blood [6, 7], urine [8, 9, 10, 11], saliva [12] and hair [13]. Usually, the concentration of amphetamines in body gradually decreases over time due to metabolism and excretion. Therefore, a sensitive, accurate, simple and rapid method is essential for drug screening in forensic toxicology.
Recently proposed methods for determination of amphetamines require that the biological samples be purified, concentrated or derived for the following GC [8], GC-MS [7, 9, 12] and HPLC [10, 11, 13] to detect. Since the molecular weights of amphetamine and methamphetamine are small, their retention time is usually short and the peak shapes are not legible. Thus, derivation is often needed to improve sensitivity on the burden of a relatively complex sample preparation. Although HPLC is traditionally another choice, its determination generally faces problems of long analysis time and severe matrix interference. The UPLC-MS/MS method is characterized with a very high sensitivity for the analytes as well as a good accuracy of quantification. This means UPLC-MS/MS alternative could detect a wide range of amphetamines simultaneously using a simple sample clean-up procedure. Here, a reliable UPLC-MS/MS method was established for the quantitative measurement of six amphetamines in urine, with an SPE extraction technique to yield cleaner analytic extracts to minimize the matrix effect and protect the chromatographic column and mass spectrometry instruments from contamination.
Obtainment from Cerilliant (Round Rock, TX) is amphetamine(AM), methamphetamine(MA), 4-Methyle-nedioxyamphetamine(MDA), 4-methylenedioxyme-thamphetamine(MDMA), N-Methyl-1-(3, 4-methyl-enedioxyphenyl)-2-butanamine(MBDB), 4-methylenedioxyethylamphetamine (MDEA) and MA-d5 at 1 mg· mL-1. All reagents are of HPLC grade. Acetonitrile (ACN) and methanol (MeOH) were purchased from Fisher Scientific (Fairlawn, NJ). Formic acid (FA) was obtained from Spectrum Chemical MFG Corp. (Gardena, CA). Deionized water was generated with Elix, a Millipore water purification system (Millipore Corp., Molscheim, France).
The analytes were separated on a column (ACQUITY UPLC BEH Phenyl column; 100 mm× 2.1 mm, 1.7 μ m) that was held at 35 ° C. The mobile phase, consisted of ACN (eluent A) and 0.3 % FA (eluent B), was flowing in gradient at a rate of 0.40 mL/min. Immediately after sample injection, the ACN percentage was increased from the initial 10 % to 55 % over 3 min, and increased to 90 % in 1 min, then decreased to 10 % in 0.1 min with a post equilibration time of 1.9 min. The total run time was 6 min.
Ionization was achieved with electrospray in the positive ionization mode (ESI+). Nitrogen was used as the nebulization and desolvatation gas. The multi-reaction monitoring mode (MRM) was used, followed by the dependent scan, which was an enhanced product-ion scan. The two resulting transitions per analyte and respective setting for UPLC-MS/MS running were given in Table 1. The other main parameters were drying gas temperature of 500 ° C, source heater temperature at 300 ° C, nebulization gas flow in 650 L· h-1, cone gas flow of 150 L· h-1 and capillary voltage as 3.0 kV.
| Table 1 Two resulting MRM transitions per analyte and corresponding settings for UPLC-ESI-MS/MS system (PI: parent ion; DI: daughter ion; CV: cone voltage; CE: collision energy) |
Methanolic standard stock solutions (1 μ g· mL-1) of each analyte were prepared from the commercially available methanolic solutions (1.0 mg· mL-1). The working standard solutions were prepared from the stock solutions by dilution to the appropriate concentrations as needed. The internal standard (IS) of MA-d5 was prepared to produce its working solution (100 ng· mL-1) in methanol. All the working solutions were stored at -20 ° C.
Human blank urine, used for development and validation of the proposed method, were obtained from volunteers without history of drug abuse, storing in refrigerator at -20 ° C.
The extraction of analytes in urine was performed with Oasis MCX solid phase extraction (SPE) cartridge (1 mL, 30 mg; Waters, USA). 2 mL of spiked urine sample (with 1 ng· mL-1 MA-d5 as internal standard) was centrifuged for 5 min at 8000× g and the supernatant was loaded into an SPE cartridge which was preconditioned by the first addition of 1 mL methanol followed by 1 mL deionized water and allowed to flow freely under gravity. The SPE cartridge was washed with 1 mL of 10 % methanol in water. The columns were dried under vacuum for 10 min, then leaving the analytes to be eluted with 2 mL of 5 % ammonia water and 70 % hydrated methanol. The elution was submitted for UPLC-MS/MS analysis.
Selectivity was evaluated by analyzing ten separate sources of blank urine for background noise and identifying matrix interferences. Mean area measurements for the selected analyte ions were generated and analyzed to ensure that the matrices were free from interfering compounds. As shown in Fig. 1, no interfering peaks were observed in the extracts of blank urine samples, indicating that the assay was found to be selective for all tested compounds.
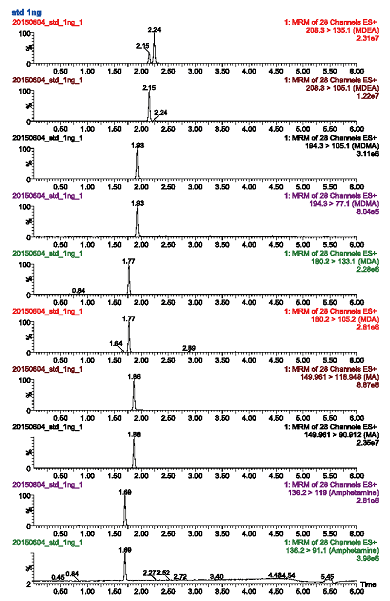 | Fig.1 MRM chromatograms of extracted urine sample solutions (at 10 ng/mL) containing the six amphetamine-type drugs |
Calibration standards (with 1 ng· mL-1 MA-d5 as internal standard) for urine samples were obtained between 0.1 to 20 ng· mL-1, depending on the sensitivity of the analytes. Daily calibration curves of the same concentration were prepared with each batch of validation and authentic samples. For all analytes, a linear weight (1/concentration) model was found to be the best and therefore used for the calibration curves. The regression equations and correlation coefficients of each analyte are listed in Table 2. The lower limit of quantification (LLOQ) and the limit of detection (LOD) in the MRM mode were defined as the concentrations with a signal-to-noise ratio of 10 and 3, respectively. The LLOQs corresponded to the lowest concentrations were used for the calibration curves with a signal-to-noise ratio of at least 10. The LODs reached 0.005~0.02 ng· mL-1 for urine samples.
| Table 2 Regression equations with correlation coefficients and LODs for the given analytes in urine samples |
The concentrations for QC samples were 0.125, 1.25 and 12.5ng· mL-1. They were selected to perform the accuracy and precision test. According to the procedure described in the preceding text, five QC samples’ replicates of low, middle and high concentration were run on three consecutive days. The concentrations of the analytes in the QC samples were calculated via the daily calibration curves. Precision for intra- and inter-day was calculated, given as relative standard deviation (RSD).
Extraction efficiencies and matrix effects were estimated in a post-extraction addition approach. Three sets of samples of five different blank matrices were prepared at three QC concentrations. Samples in set 1 consisted of neat standards. For preparation of the samples in set 2, blank urine samples were first extracted, as described previously. Then, the extracts were spiked with corresponding working standards and the IS. For preparation of set 3, blank urine samples as in set 2 were spiked with corresponding working standards and the IS at three concentrations. Thereafter, they were extracted through the previously-described extraction procedure. Extraction efficiencies were estimated by a comparison of the peak areas from the samples of set 3 with those from the corresponding samples of set 2. Matrix effects were estimated by a comparison of the peak areas from the samples of set 2 with those from the corresponding samples of set 1. Data were listed in Table 3. Intra- and inter-day precision was lay within the required limits of 15 % RSD (20% RSD at LLOQ) at all studied concentration levels. Process efficiencies were satisfying and matrix effects were in a non-disturbing range.
For estimation of stability of the processed samples under the conditions of UPLC-MS/MS analysis, three concentration levels of QC samples (n=3/each) were extracted. The resulting extracts at each concentration level were maintained at room temperature (+18 ° C) for 24 h before analysis. For the evaluation of stability after three freeze-thaw cycles, the QC samples were frozen at -20 ° C for 24 h. After this period, the samples were again thawed and frozen for 24 h and this process was repeated until the third thawing cycle when the QC samples were extracted and analyzed. The concentrations of the extracted analytes in the QC samples were calculated via the daily calibration curves. Stability of the analytes demonstrated that there were no significant differences from the reference concentrations.
The developed method was applied in real forensic cases. These cases involved people who had drug history. 58 urine samples were collected from different sources and diluted by 1000 times. MA was detected in all of the 58 urine samples. The measured concentrations were listed in Table 4. Results indicated that the developed method was suitable for drug screening in forensic case.
| Table 3 Intra- and inter-day precision, extraction efficiency and matrix effect (values given in %) of the UPLC-ESI-MS/MS assay for each analyte (n=5) |
| Table 4 The measured concentration of MA in 58 urine samples by the proposed method |
A rapid and sensitive method for simultaneous determination of six amphetamine-type drugs in urine was developed and validated through purification by Oasis MCXSPE cartridge and detection by UPLC-MS/MS. The detection sensitivity was improved significantly in comparison to the other choices. The method was proved to be a reliable alternative for determination of amount-trace analytes in urine.
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|