{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
三个姓氏聚居村家系Y-STR单倍型分析
[刘宏1  , 金国文
, 金国文2 , 杨幸怡1 , 李越1 , 苏会芳1 , 刘超1, * ]
, 金国文]
|
|


作者简介: 刘宏,副主任法医师,硕士,研究方向为法医遗传学。 E-mail: 79liuhong@163.com
目的利用Y-STR单倍型建库进行家系排查,对相对封闭的姓氏聚居区域发生的案件,或以姓氏亲缘为纽带的团伙性案件的侦破,具有其它技术不可替代的作用。近年来,国内已有相关报道与实际应用,但针对姓氏聚居区域Y-STR单倍型分布特点的基础研究缺乏,Y-STR家系数据库建设与家系比对排查也缺少理论支撑。本文通过对3个聚居姓氏家系样本的Y-STR单倍型检测分析,探讨如何更好地使用Y-STR单倍型做家系排查及数据库建设。方法 以姓氏聚居的自然村为研究区域,以五代父系亲缘为一个家系单位,按区域内该姓氏家系总数1/5随机选取家系,采集该家系内全部男性个体样本,共获3个姓氏72个家系426份样本。其中,河北廊坊市某镇某村苏姓30个家系155份样本,杨姓20个家系92份样本;广西岑溪市某村林姓22个家系179份样本。应用Y-filerTM试剂盒对上述样本进行Y-STR单倍型检测,并对获得的单倍型进行分析。结果 检测的3个姓氏72个家系426个体的Y-filer系统,共获得46个单倍型,各单倍型所包含的个体数量在群体中分布不均衡,并且根据差异大小呈“集合”分布。其中,苏姓30个家系中共有10个单倍型,分为5个单倍型集合;杨姓20个家系中共有14个单倍型,分为两个集合;林姓22个家系中共有22个单倍型,形成4个集合。不同姓氏间未见相同的单倍型。如根据单倍型在同姓中的分布特点,将同源集合里包含最多家系数的代表性单倍型定义为主流单倍型,以主流单倍型作为该姓氏Y-STR的“标识物”,则苏姓中的H1,杨姓H11、H12、H17和林姓H25、H36为主流单倍型。以主流单倍型作为比对对象,基因座容差范围设置为2,可覆盖122名苏姓、59名杨姓和130名林姓人员,取样群体占比分别为78.7%、64.1%、72.6%。72个家系的17个Y-STR基因座共观察到19次突变,其中苏姓30个家系共113次父子遗传,2个家系共发生3次突变,平均突变率为0.001 6 (95%CI,0.000 3~0.004 6);杨姓20个家系共有69次父子遗传,6个家系共发生7次突变,平均突变率为0.006 0(95%CI,0.002 4~0.012 3);林姓22个家系中共有152次父子遗传,9个家系共发生9次突变,平均突变率为0.003 4(95%CI,0.001 7~0.006 3)。3个姓氏所有个体中,17个Y-STR基因座平均突变率为0.002 3 (95%CI,0.001 2~0.005 1),突变基因座分别为DYS392、DYS437各1次;DYS390、DYS635、DYS439各2次;DYS389II 3次;DYS458、DYS385a/b各4次,增加突变8次,减少突变11次,1步突变18次,2步突变1次。结论 本研究显示同姓聚集地的不同单倍型可形成同源集合,验证了姓氏与Y-STR的密切关联,同一姓氏内家系由于入赘、改姓等非遗传原因会造成差异。利用Y-STR单倍型进行家系排查,考虑突变现象,可根据具体情况设定与主流单倍型的容差数,如嫌疑人的单倍型有1~2个Y-STR基因座分型与主流单倍型不同时,不宜轻易改变排查范围,应结合调查筛选重点个体或家系,补充Y-STR基因座检测比对,结合常染色体STR检验比对锁定嫌疑人。此外,还要考虑Y-STR的突变规律,引入差异步长作为比对条件,如差异为高突变基因座且差异步长为1,需增加Y-STR基因座检测数目。姓氏Y-STR数据库应以自然村(组)为单位确定“主流单倍型”,然后由近及远作单倍型并类分析,逐步确定村、街、镇姓氏主流单倍型。从投入、效益考虑,Y-STR建库主体应以地级市为主,按人口姓氏、犯罪群体分布特点,确定那些区域、哪些姓氏优先建库。地级市库尽快实现远程自由查询比对,以缩小嫌疑人排查范围。省级和国家级Y-STR数据库应根据犯罪类别,主流单倍型代表的群体和人口数目筛选入库,自动比对和发布。Y-STR排查和数据库建设采样策略是通过采集有限的样本获得该姓氏最具代表性的“主流单倍型”,以其作基准数据,覆盖最多姓氏成员。建议家系Y-STR样本采样方案为:“长辈优先,平衡采样,三代必采”。“长辈优先”是指辈分高者优先采样,辈分高的成员拥有家系中最大的共性Y-STR单倍型,距离其他成员间的平均遗传距离最短,产生突变的几率最小;“平衡采样”,即谱系同辈的不同分支间,同代采样数量应根据该分支的男性个体数平衡采集,目的是有利于确定Y-STR主流单倍型,同时可以评估不同分支内是否存在Y-STR突变;“三代必采”,是指不能漏掉某一分支,每三代内至少采集一个样本,以获得更加完整的家系Y-STR,以发现不属同一家系的Y-STR单倍型。总之,运用Y-STR进行排查的准确性与该姓氏起源、家系迁徙、遗传突变、人口数量、采样比例等都有关系,有时即使选择数个主流单倍型也难以覆盖区域内全部姓氏人员。
Author: LIU Hong, master, associate chief forensic physician in forensic DNA test and research. Email: 79liuhong@163.com
* 通讯作者: 刘超,主任法医师,博士生导师,博士,研究方向为法医遗传学。 E-mail: liuchaogzf@163.com
*Corresponding Author: LIU Chao, Ph.D, chief forensic physician and instructor of doctoral candidates. Email: liuchaogzf@163.com
Objective To investigate into the screening, pedigree identification and database construction of Y-STR’s main haplotypes. Methods Haplotypes at 17 Y-STR gene loci of 426 males from 72 pedigrees having descended beyond five generations in three surname-concentrated villages were surveyed and analyzed with Y-filerTM kit, and the number of haplotypes was counted along with the coverage rate of main haplotypes. Results A total of 46 haplotypes were detected at 17 Y-STR gene loci from the 72 pedigrees investigated, with each surname’s pedigrees having about 1 or 2 main haplotype(s) which were originated to form the other mutated or varied ones. The population coverage rate was 62%~79% when the fault allowance for variance set at two gene loci. Conclusions Database of Y-STR’s main haplotypes by surnamed-pedigree, once set up, could help to reduce the scope to find suspects, target at certain pedigree, though inability to directly identify the suspects from a same surname-concentrated community.
It is crucial to set up Y-STR haplotypes database of regional pedigrees to orient a criminal suspect to certain pedigree when either the crime happens in some surname-concentrated communities of consanguinity and low migration or the suspect’ s Y-STR haplotypes link with a certain surnamed pedigree whose members involve in gang-crimes [1]. Therefore, in recent years some domestic forensic institutions have attempted to develop Y-STR databases of pedigrees, yet lacking the fundamental research on distribution characteristics of Y-STR haplotypes in surname-concentrated communities, thereby resulting in less theoretical support for both the establishment of Y-STR database of pedigree and the methods for pedigree comparison [2]. Through detection and analysis of Y-STR haplotypes from pedigree specimens in surname-concentrated natural villages, we explored how to correctly use Y-STR haplotypes with purpose to help the pedigree identification and relevant database construction.
Families of certain surnames, having complete and clear genetic lineage, were selected from surname-concentrated natural villages. With five generations of lineal kin as a pedigree unit and one fifth of the total population of the investigated pedigree as the sampling size, the pedigree was selected randomly and all the males sampled were of informed consent. There were 426 males from 72 pedigrees of three surnames, among which 155 from 30 pedigrees of surname Su and 92 from 20 pedigrees of surname Yang, each located in an individual village of Langfang city, Hebei province, were chosen along with the other 179 ones from 22 pedigrees of surname Lin, living in a village of Cenxi city, Guangxi Zhuang autonomous region.
All specimens were extracted with paramagnetic beads method on Freedom EVOTM full automatic workstation platform (TECAN, Switzerland). The magnetic beads used were from EQ1000TM extraction kit for forensic medicine (Eastwin Life Sciences Inc, China) [3].
DNA from all specimens was amplified and typed through AmpFLSTR ® Y-filer TM kit (Life Technologies Corporation, UK) according to related literature [4].
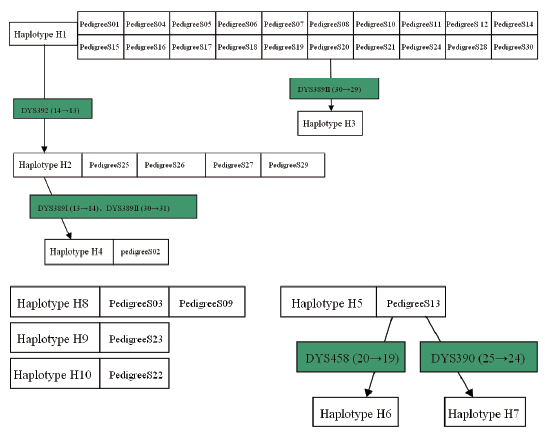
A total of 46 haplotypes at 17 Y-STR gene loci were detected from 426 individuals of 72 pedigrees investigated. Amid them, 10 haplotypes were from 30 pedigrees of surname Su, 14 haplotypes were from 20 pedigrees of surname Yang, and 22 haplotypes existed in 22 pedigrees of surname Lin, with no identical haplotypes found among different surnames.
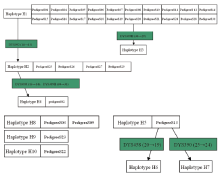
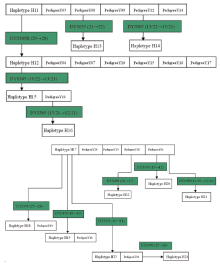
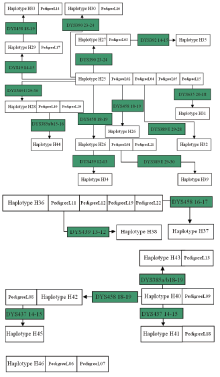
It was shown that different conglomeration of Y-STR haplotypes emerged amidst the same surname’ s diverse haplotypes, as indicated in Fig.1~3 revealing the relationship of various haplotypes observed from surnames Su, Yang and Lin, respectively.
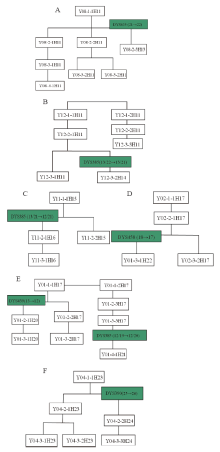
 | Fig.1 Main haplotypes and its branches of Su-surnamed pedigree |
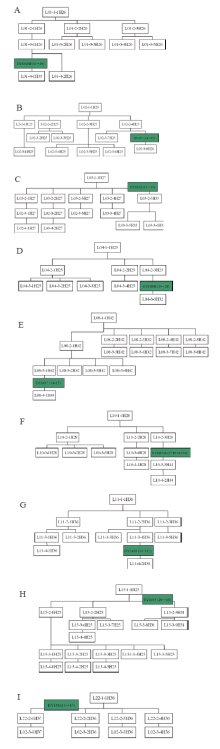
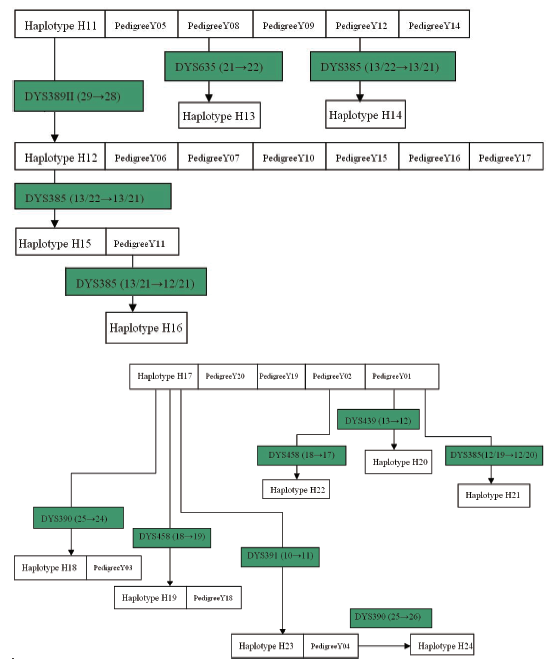 | Fig.2 Main haplotypes and its branches of Yang-surnamed pedigree |
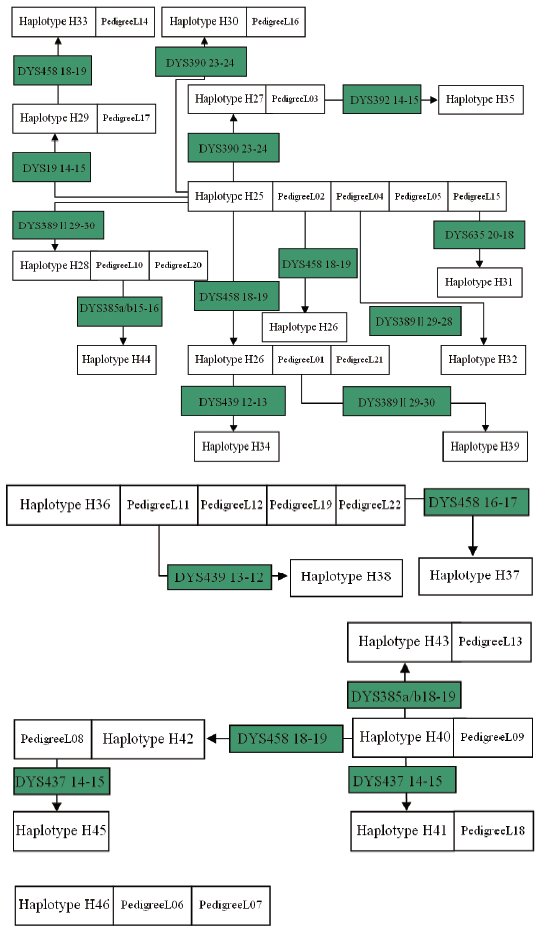 | Fig.3 Main haplotypes and its branches of Lin-surnamed pedigree |
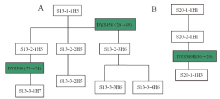
In the 30 pedigrees of surname Su, the descendibility from father to son occurred 113 times with 2 pedigrees having happened 3 mutations and the average mutation rate of 17 Y-STR gene loci being 0.0016 (95%CI, 0.0003~0.0046). For the 20 and 22 pedigrees of surnames Yang and Lin, the same descendibility was 69 and 152 times, mutations having 7 and 9 pieces in each of 6 and 9 pedigrees, together with the average mutation rate of 17 Y-STR gene loci to be 0.0060 (95%CI, 0.0024~0.0123) and 0.0034 (95%CI, 0.0017~0.0063), respectively. All tested males from the three surnames got the average mutation rate of 17 Y-STR gene loci as 0.0023 (95%CI, 0.0012~0.0051) and the mutation gene loci were DYS392 and DYS437 (1 time for each); DYS390, DYS635 and DYS439 (2 times for each); DY-S389II (3 times); DYS458 and DYS385a/b (4 times for each). There were also 8 added yet 11 reduced repetitions in different places. The pedigrees with Y-STR mutations were shown in Fig.4~6.
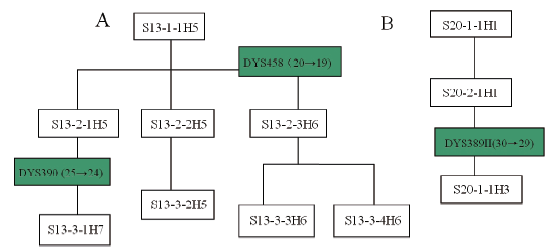 | Fig.4 Y-STR genetic genealogy of the surname Su’ s pedigrees carrying mutations. A: pedigree 13; B: pedigree 20. For S13-1-1H5 to be taken as indication of the symbols, S represents surname Su, 13 the serial number of the investigated pedigree, 1 the generation grade the sample resides at the pedigree, the followed 1 the given number. of the sample, and the final H5 the haplotype. |
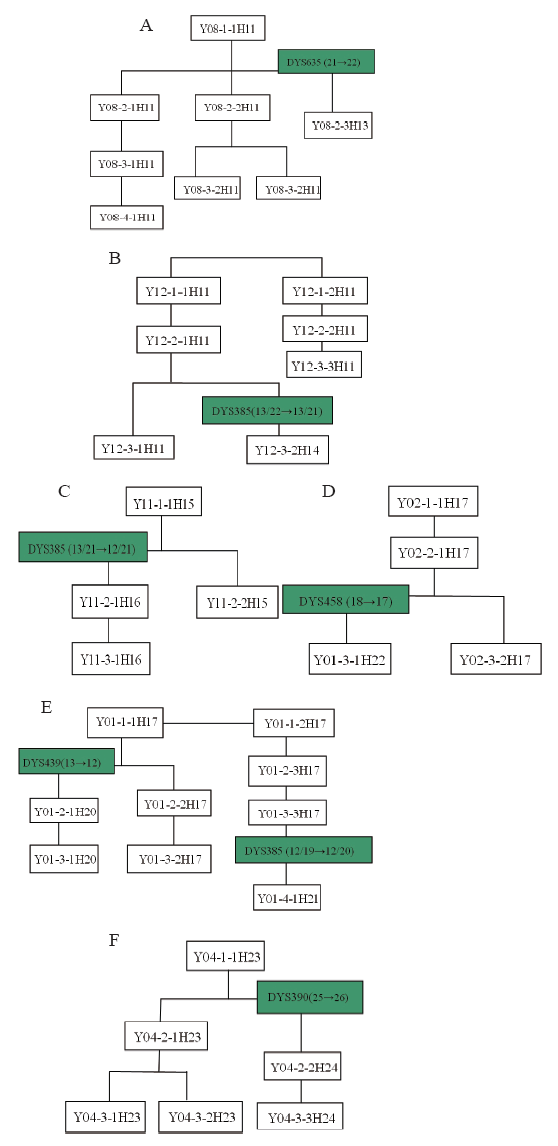 | Fig.5 Y-STR genetic genealogy of the surname Yang’ s pedigrees carrying mutations (Symbols’ meaning is same as indicated in Fig.4) |
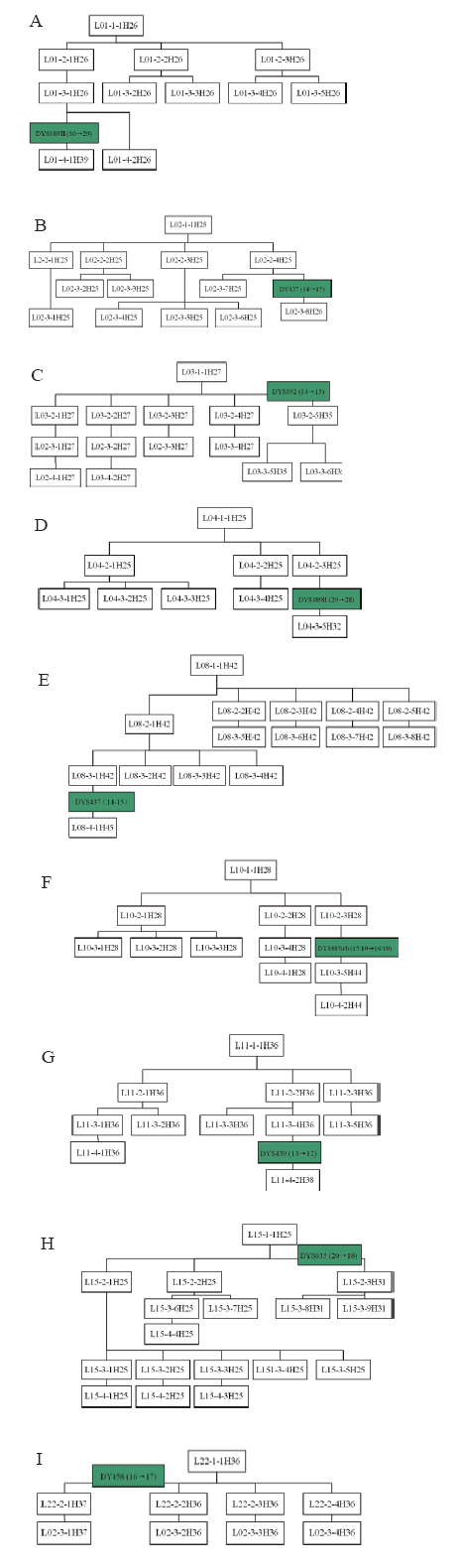 | Fig.6 Y-STR genetic genealogy of the surname Lin’ s pedigrees carrying mutations (Symbols’ meaning is same as indicated in Fig.4) |
STR gene loci on Y chromosome, characteristics of heredity by patrilineal haplotype, bring about the descendants from same paternal line to carry the identical Y-STR haplotypes except for gene mutation. It is a long-set tradition for Chinese to inherit their paternal surnames, leading to their origins likely to be traced with the help of Y-STR linked to certain surnames. Because of the similarity for Y-STR and surname to be passed down, population genetic and anthropologic researches take as tools of Y chromosome as the inner or biological hereditary factor and surname as the external or social manifestation.
It was observed in this study that several homologous conglomerations were formed from various haplotypes which were carried with pedigrees of same surname-concentrated community, in consistency with the intimateness between surname and Y chromosome. For example, five sets of conglomeration were formed out of 10 haplotypes in 30 pedigrees of surname Su as shown in Fig.1, with one set consisted of H1, H2, H3 and H4, the second set of H5, H6 and H7, and each of H8, H9 and H10 into its own separate set. Haplotype H1 of pedigree 20 took place a mutation in its DYS389II (30) when passed down from father to son, resulting in the son bearing DYS389II (29) and haplotype H3 being formed. Haplotype H2 existing in 4 pedigrees was only different from H1 at the DYS392 where H1 was 14 yet 13 for H2. H2 should be from the same patriline with H1 and formed over mutation. Haplotype H4 owned in one pedigree, having discrepancy of just one repetitious unit at the each allele of DYS389I, DYS389II, DYS392 against the corresponding one of H1 along with H2’ s DYS389I, DYS389II, is very likely from the same patriline as H1 and H2, with H2 likely the more closer, because DYS389 is a composite gene locus whose sequence encloses DYS389I and DYS389II so that DYS389I, once mutated, will make DYS389II follow to change. Therefore, the pedigrees containing H1, H2, H3 and H4 can be classified into one set of conglomeration based on the less-discrepant degree and relationship among them. H5, owned with pedigree 13 only, has 11 different genotypes of loci from those of H1, keeping it unable to be in same set with H1. A mutation suffered to a male in the pedigree at the locus DYS458 (20→ 19) made H6 born and the other one happening in another male at the DYS380 (25→ 24), having caused the successor having H7, thus, H5, H6 and H7 belong to one same set. For H8, H9 and H10, not only are they completely independent because of their far-distant relationship but also have their respective 11, 9 and 11 dissimilar genotypes of loci in contrast to those of H1, the huge distinction bringing each of them into disparate set against H1. As such, H8, H9 and H10 can only be collected into their own individual set.
As for surname Yang, whose pedigrees formed two sets of conglomerated haplotype as shown in Fig.2 where haplotype H11 is shared with 5 pedigrees, out of which the pedigree 08 underwent a mutation on DYS635 (21→ 22) to let the haplotype H13 come into being in a new branch that contains only one person for this survey. Similarly, haplotype H14 was born from the same way as H13 when a mutation occurred on DYS385 (13/22→ 13/21) of pedigree 12. H14 is also owned with only one male for this investigation. The naissance of H12 is due to a mutation happening on H11’ s DYS389II (29→ 28), with the resultant branch consisting of 6 pedigrees. Unfortunately, the generation of what H11-bearing pedigree to produce haplotype H12 has not been known because of lack of materials and information. Pedigree 11 holds haplotype H15, deduced to be born from the mutation on H12’ s DYS385 (13/22→ 13/21). When a mutation on DYS385 (13/21→ 12/21) occurred on the passing down from a father to his son, the H16 emerged. Therefore, H12, H13, H14, H15, H16 are all branches of H11, the pedigrees of H11, H12, H13, H14, H15 and H16 should be collected into a same haplotype clan. H17 is another set of conglomerated haplotype of surname Yang, shared with four pedigrees of 01, 02, 19 and 20. A male of pedigree 01 came about a mutation on DYS439 (13→ 12) to bring the birth of H20 (detected and verified from the man’ s son and grandson), and the mutation on DYS385 (12/19→ 12/20) of another man conduced the emergence of H21 (carried by the son). Haplotype H22 arose from the mutation on DYS458 (18→ 17) of a person in pedigree 02 (son is the carrier). H18 in pedigree 03 was formed when H17’ s DYS390 mutation (25→ 24) took place. Likewise, mutations on H17’ s DYS458 (18→ 19) and DYS391 (10→ 11) gave birth of H19 in pedigree 18 and H23 in pedigree 04 where a man’ s DYS390 mutated (25→ 26) to deliver H24 (detected and verified from the next generations). In summary, H18, H19 and H23 all came from H17’ s mutation although the updated materials till present have not traced which generation of what pedigree to produce the mutations. In a word, pedigrees from H17 and its branches H18, H19, H20, H21, H22, H23 and H24 are in one set of conglomerated haplotype.
Pedigrees of surname Lin’ s haplotypes formed four sets of conglomerated haplotype as shown in Fig.3. Among them, H25, shared with 5 pedigrees of most population, is by the difference of 1~2 Y-STR gene loci to cause birth of a total of 13 conglomerated haplotypes including H26, H27, H28, H29, H30, H31, H32, H33, H34, H35, H39 and H44. Also with the difference of 1~2 Y-STR gene loci, haplotype H36 delivered H37, H38 to constitute their group of 3 sorts of haplotype; H41 produced H40, H42, H43 and H45 to make up of their group of 5 kinds of haplotype. Yet H46 is a separate conglomeration, owned with an individual pedigree of 10 people.
Based on the distribution characteristics of haplotypes among pedigrees of same surname, a main haplotype is defined as the representative haplotype that is shared with most individuals of a homologous conglomeration. The main haplotype stands for a marker of Y-STR gene loci of certain family. Therefore, H1 is the main haplotype of surname Su’ s pedigrees, the three of H11, H12 and H17 are of Yang’ s, and Lin’ s of H25 and H36.
Gene mutation is very influential on Y-STR heredity. Upon our previous observation on 17 Y-STR gene loci to determine the paternity from 1, 000 pairs of father and son, there were 46 mutations found in 11 gene loci, with the average rate of mutation on the 17 Y-STR loci to be 0.0027 (95%CI, 0.0020~0.0360) [5]. This investigation showed that 7 mutations occurred in the 6 pedigrees out of total 20 ones of surname Yang together with the average mutation rate of the 17 Y-STR loci as 0.0060 (95%CI, 0.0024~0.0123), having verified the higher mutation rate of Y-STR haplotypes distributing in the pedigrees of same surname, too. Many haplotypes have difference of 1 or 2 gene loci from the relevant main haplotype, leading to be thought as the branch haplotypes from mutation of the main haplotype. Therefore, with quantity less than 2 for either the mutation times or gene loci involved in mutation in this investigation, such haplotypes were put under their respective same main haplotypes, in accordance with the viewpoint of Wu et al [6]. With fault allowance for variance set within 2 gene loci to identify the relevant ones by main haplotype, 122 individuals from surname Su were enclosed, accounting for 78.7% of the total sampled population. For those of surname Yang and Lin, their individual amount was 59 and 130, and the percentage 64.1% and 72.6%, respectively. If the fault allowance for variance set within 3 gene loci, the pedigrees will be covered much greater. In practical use, the quantity of fault allowance can be set based upon concrete conditions. As supplementary means to identify or exclude certain population in specific area, Y-STR screening and the above handling often play enormous significance.
From two aspects comes the difference of Y-STR haplotypes among various pedigrees of a same-surname concentrated community. One is the relevant pedigrees did not originate from an identical ancestor, thus making a comparatively larger diversity of Y-STR so that the unit of core sequence of many gene loci repeats more than two times, a phenomenon unable to explain with mutation [5]. The other one arises from the Y-STR mutations occurring in the pedigrees of same patriline to cause difference. Such disparity behaves as the haplotypes collected from their conglomeration, with small amount of diverse gene loci (generally only 1~2 gene loci) and the quantity gap of repetitious alleles just at 1 or 2 pieces, all of above in conformity with the general rule of STR mutation to give the explanation for their genesis. Most of the Y-STR mutations are of one step, but the mutations within three steps or mutated gene loci no more than three pieces between two individuals are likely to happen in the course of descending from father to son, suggestive of the male individuals owning these haplotypes likely coming from same patriline. Evidently, when Y-STR haplotypes are used to identify or exclude pedigrees and there are differences of 1 or 2 Y-STR gene loci of the suspect from the main haplotype, mutations should be considered and the scope of exclusion cannot be changed incautiously. The detection and matching of extra Y-STR jointed with euchromosomal STR ought to conduct to ascertain the suspect in assistance of the investigated and screened key individuals and/or pedigree. Presently it has been ordinarily accepted that the criterion on Y-STR identification refers to that on paternity exclusion of euchromosomal STR that presence of 3 different gene loci can exclude paternity [7]. Whereas, our previous research demonstrated that mutation rate of relevant Y-STR gene loci should be thought even if there were 3 different genotypes of relevant Y-STR gene loci present between the suspected father and son. On existence of three gene loci of high mutation, the best way is to add the amount of detected Y-STR gene loci so as to make a safe exclusion with the basis of one more new mutated gene loci being discovered.
The establishment of Y-STR database of pedigrees dominated by surname should be carried out as follows: to begin with is counting the main haplotypes of certain surnamed pedigrees in a natural village (or group), as the minimal targeted unit, and classifying the discovered haplotypes based on near-to-far relativeness, the subsequent is gradually extending to thorp, street communities, town for keeping records of the counted haplotypes of relevant surnames [2]. With consideration on investment and efficiency, criterion must be emphasized on the first priority and database establishment be conducted at different levels. For the prefecture-scale cities to be as dominant centers, which surname and what area can be preferentially selected to set up the database should be stood on the surname’ s distribution in major population and the characteristics of criminals scattering. The prefectural database can be executed the unrestricted remote-distance query and match at the disposal of authority, so as to reduce the size of identification and exclusion. Provincial and national Y-STR database should be founded on the crime types and the screened-out main haplotypes representative of population and amount [8]. For better service to actual combating against criminals, it is also necessary to develop the reagents including more Y-STR gene loci labeled with fluorescence of 5 or 6 colors and/or concurrent autosomal STRs, able to make more information acquired with one-time detection [9].
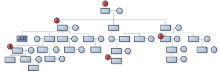
The aim to set up Y-STR database dominated by surname is, on inability to know the mutations, acquiring the most-shared preponderant Y-STR haplotypes from limited sampled specimens of certain surname with their dominance, thereby being as the benchmark to produce the maximal data-matching to cover most members of the relevant surname’ s pedigrees. To achieve the goal, the priority is how to determine the sampled subjects selected for database establishing. From mutation traits of the genetic profiles obtained in this study on the relevant pedigrees, the principle on specimen-sampling to set up Y-STR database of pedigrees dominated by specific surnames can be concluded that: A) placing priority onto first generation; B) assuring sampling in balance, and C) of every three generations must be collected (at least) one sample. For A, first generation, the member of the eldest and highest hereditary rank in the pedigrees, will be on top priority to be sampled because the Y-STRs descend from the members of higher hereditary rank to the ones of the lower, and members of highest hereditary rank carry the largest and quantity-maximal most-shared Y-STR gene loci that are least heredity-distant from else members and smallest probability of mutation. For B, sampling in balance indicates that sampling should not overweigh on certain pedigree or its some branch or members but evenly collect samples from diverse pedigrees and respective hereditary compeers, thereby harvesting the most-shared main haplotypes by which to assess the presence of Y-STR mutations in different branches of the pedigree so as to attain more accurate Y-STR pedigree pattern. About C, a sample must be picked up at and within the interval of every three generations on conditions permitted, thus making up the validity and integrity of relevant pedigree’ s Y-STR model. The above principle was illustrated in Fig.7.
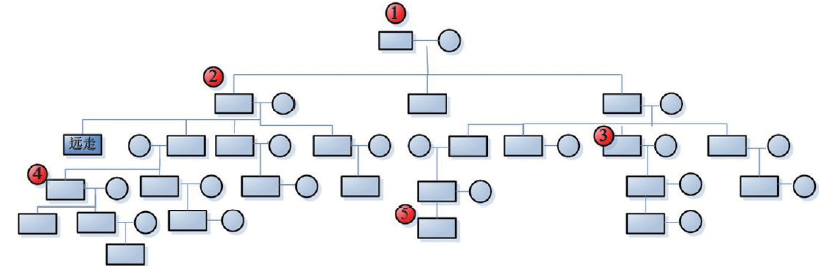 | Fig.7 Schematic diagram for principle on sampling large pedigree. The number in the figure represents the priority of sampling. |
Notably, pedigree comparison by Y-STR could be influenced by various factors like the history of surnamed pedigree, the origin of a pedigree, mutations, sampling ratio and other random elements. Sometimes, it is difficult to cover the whole population of certain surname in an area with selection of several main haplotypes.
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|