{kind=link}
{kind=link}
二代测序技术及其在法医遗传学中的应用
[王乐 , 叶健, 白雪, 杨帆, 赵兴春
, 叶健, 白雪, 杨帆, 赵兴春* ]
, 叶健, 白雪, 杨帆, 赵兴春]
|
|


作者简介: 王 乐(1983—),男,辽宁沈阳人,副主任法医师,博士,研究方向为法医遗传学。 E-mail: wangle_02@163.com
从DNA指纹图谱到STR复合扩增检验,30年来法医DNA工作者目睹着技术的深刻变革以及在此推动下案件侦查模式的巨大转变。自从二代测序技术问世以来,遗传学的研究方式已发生了巨大转变。但是相比其在癌症和遗传病诊断、基因组从头测序和重测序、转录组重测序、药物研制等领域的应用,二代测序在法庭科学领域的应用尚处于起步阶段。本文介绍二代测序的基本概念、发展历史和工作原理,综述了二代测序技术在STR分型、SNP分型、线粒体全基因组测序等几个重点领域近两年的最近动态,最后结合二代测序在法庭科学领域的应用展望提出可能遇到的挑战,希冀对相关研究和实践提供参考。
From DNA fingerprinting to multiplex STR amplification and detection, forensic DNA scientists witnessed the rapid advances in DNA technology and the substantial changes in ways of solving criminal cases during the past three decades. As a matter of fact, only incremental developments of forensic DNA technologies and the "passive comparison" mode of using DNA information could not meet current expectations for forensic genetics from crime investigators. It has been unprecedentedly emphasized that great efforts are needed for more powerful solutions that are automatic, high-throughput, precise, rapid and being support to the "active searching" mode of DNA information utilization. Under such circumstances, next generation sequencing (NGS) comes just in time. Chinese authorities and experts have already realized the great potential of NGS applications for forensic purposes, although the application of NGS in forensic science is still at initial stages, compared with its applications in fields of cancer diagnosis, genetic disease diagnosis, de novo sequencing, genome resequencing, transcriptome resequencing and drug discovery. More information can be obtained from a single experiment by analyzing the STR, SNP, Indel and RNA markers simultaneously, which could be impossible on routinely used PCR-CE platforms because of the limited amount of exhibits. In this article, the authors attempt to describe the basic concepts, developmental history and working principles of NGS to Chinese experts in the general field of forensic science and technologies, and share the updates of NGS-based STR typing, SNP typing and whole mtGenome sequencing during the past two years. Representative NGS platforms including the 454/Roche GS FLX system, the Solexa system, the SOLiDTM system, the Ion PGMTM system and the MiSeq FGxTM system were introduced. Annual statistics of research articles on NGS and forensic NGS were described and trends for related research were analyzed. Finally, perspectives of forensic NGS were presented and possible challenges including data analysis methods, openness of NGS systems and ethical issues were discussed in the hope of providing a reference for related research and applications.
自从二代测序(second generation sequencing, SGS)技术和平台问世以来, 遗传学的研究方式已发生了巨大转变。如今, 每周都有基因组数据发表, 测序速度越来越快, 测序成本越来越低。在过去十年间, 二代测序的方法和平台变得日趋完善, 数据准确性显著提升, 并开始运用于人类临床诊断。法医遗传学领域也开始关注二代测序, 在科技论文发表和学术会议报告方面, 二代测序相关成果数量呈现爆发式增长, 这些成果为解决刑事案件提供新的可能性。运用二代测序技术, 基于一次实验、一份微量生物样本检材就可以同时获得短串联重复序列多态性(short tandem repeat, STR)、单核苷酸多态性(single nucleotide polymorphism, SNP)、插入缺失多态性(insertion/deletion polymorphism, Indel)、mRNA等各种类型的大量的遗传标记信息, 这是现有PCR-CE(PCR-capillary electrophoresis)平台所无法做到的。运用二代测序技术, STR等位基因间精细的序列差异得以彰显, 未知的稀有STR等位基因得以被发现, 这些详细的序列信息有可能辅助混合样本的数据解析。本文将简单介绍二代测序的基本概念、发展历史、工作原理, 并对其在STR分型、SNP分型、线粒体全基因组测序领域的最新进展进行综述。
1980年, 英国生物化学家Frederick Sanger与美国生物化学家Walter Gilbert因建立DNA测序技术获得诺贝尔化学奖。在Sanger测序中, 核苷酸扩增从某一固定碱基开始, 通过掺入双脱氧核苷酸在随机碱基终止, 通过A、T、C、G四种双脱氧核苷酸分别标记着不同颜色的荧光基团, 判定DNA序列每个碱基位置的核苷酸种类[1]。由于Sanger测序巧妙地引入了双脱氧核苷酸, 该方法也被称为双脱氧链终止法。二代测序技术出现后, 为了便于区分, 以Sanger测序为代表的DNA测序方法被称为一代测序。Sanger测序被誉为生物化学领域最伟大的发明之一, 垄断DNA测序行业三十年, 为生物科技的迅猛发展奠定了坚实的基础, 至今仍是DNA测序的主流技术。
二代测序, 也叫下一代测序(next generation sequencing, NGS)或大规模平行测序(massively parallel sequencing, MPS), 它不是DNA测序的一种方法, 而是具有共同本质属性的一类方法, 这个共同的本质属性是“ 大规模平行” , 这也正是二代测序区别于一代测序的关键所在。无论一代测序或是二代测序, 其实验结果都是由A、T、C、G 4种核苷酸组成的DNA序列。二代测序实验过程大体可分为样本准备、文库构建、测序反应和数据分析四个步骤。各公司提供的二代测序平台在测序原理方面千差万别, 但基本都体现以上实验步骤。
二代测序技术的发展进程是新型二代测序平台不断涌现、速度不断提升、成本不断降低的过程。以下就几款较为经典的二代测序平台简要介绍其工作原理。
2004年, 454生命科学公司(2007年被Roche公司收购)生产了商品化二代测序仪— 基因组测序仪20 (GS 20)[2]。2006年12月, 推出新款二代测序仪— 基因组测序仪FLX (GS FLX)[3, 4, 5]。GS FLX系统首先将基因组DNA打碎成300~800 bp片段, 在单链DNA的3’ 和5’ 端分别连上不同的接头, 每条带有接头的单链DNA被固定在一颗磁珠上, 随后扩增试剂将磁珠乳化, 形成油包水的混合物, 即形成许多个只包含一个磁珠和一个独特片段的微反应器。每个片段在自己的微反应器里进行独立扩增, 扩增产物仍然结合在磁珠上。紧接着, 携带DNA的磁珠被放入PTP板中进行测序, 板上小孔的直径决定了每个小孔只能容纳一颗磁珠, 四种碱基依照T、A、C、G的顺序依次循环进入PTP板, 每次只进入一个碱基, 如果发生碱基配对, 就会释放出一个焦磷酸, 这个焦磷酸在酶的作用下转化为光信号, 并实时地被CCD捕获, 由此可以准确、快速的确定待测模板的碱基序列。454系统是二代测序技术的代表性平台, 虽已宣布停产, 但时至今日基于该系统的研究成果仍在不断涌现。
2006年, Illumina公司收购Solexa公司, 于2007年推出二代测序仪Illumina genome analyzer[6, 7, 8]。Solexa系统的核心思想是边合成边测序。首先将基因组DNA打碎成100~200 bp的小片段, 在小片段两端加上接头, 单链DNA片段的一端通过接头与芯片表面的引物碱基互补而被固定, 另一端随机和附近的另外一个引物互补, 形成桥状结构。扩增后, DNA单分子成为单克隆的DNA簇。合成反应中, 加入DNA聚合酶、荧光标记脱氧核糖核苷三磷酸(deoxynucleotide triphosphate, dNTP)和接头引物进行扩增, 在DNA簇延伸互补链时, 每加入一个荧光标记dNTP就能释放出相应荧光, 测序仪通过捕获荧光信号即可获得待测片段的序列信息。
2007年, ABI公司推出二代测序平台— SOLiDTM系统[9]。SOLiDTM是“ 通过寡核苷酸连接和检测测序(sequencing by oligonucleotide ligation and detection)” 的英文缩写, 与GS FLX系统和Solexa系统不同, SOLiDTM系统采用的不是合成法测序, 而是连接法测序。完成文库构建后, SOLiDTM系统采用与454技术类似的乳液PCR对短片段进行扩增, 扩增产物同样固定于磁珠表面, 磁珠共价结合于玻片上。SOLiDTM系统的独特之处在于使用DNA连接酶和荧光标记寡核苷酸探针(8个碱基)实现测序反应, 并通过“ 双碱基编码” 系统实现荧光颜色识别。
2010年, Life Technologies公司收购Ion Torrent公司, 并迅速推出首款半导体测序仪— Ion PGMTM系统[10, 11]。2012年, 推出新型台式测序仪— Ion ProtonTM系统[12]。Ion PGMTM和Ion ProtonTM系统开启了后光学测序时代, 即整个测序过程不涉及光信号, 直接监测测序反应过程中氢离子释放导致的局部pH值变化, 利用离子传感器直接将化学信号转化为数字信号。2014年, Thermo Fisher公司收购Life Technologies公司, 推出两款为Ion PGMTM系统设计的法医SNP分型试剂盒:包含124个常染色体SNP位点和34个Y-SNP位点的人类个体识别试剂盒以及包含165个常染色体SNP位点的祖先来源推断试剂盒, Thermo Fisher正致力于研发CODIS核心基因座的STR分型试剂盒, 先期适用版本包含10个基因座[13]。
2014年, Illumina公司推出基于二代测序技术的法医基因组分析系统— MiSeq FGxTM系统, 并配套二代测序试剂盒— Forenseq DNA Signature Prep试剂盒[13]。MiSeq FGxTM系统继承Solexa系统边合成边测序的基本原理, 支持STR、SNP、线粒体DNA等多种法庭科学分子标记检测与分析; Forenseq DNA Signature Prep试剂盒包含27个常染色体STR基因座、24个Y-STR基因座、7个X-STR基因座、94个身源识别SNP基因座、22个表型SNP基因座和56个地域祖先来源SNP基因座。
科研论文的发表数量和变化趋势可以直观地体现某个研究领域的研究热度和发展趋势。截止2015年7月10日, Pubmed搜索引擎共收录16423篇二代测序相关科研论文, 主要集中于二代测序技术在各类癌症和遗传病诊断、基因组从头测序和重测序、药物研制、转录组、微生物组、基因调控等领域的研究和应用。自2007年起论文数量呈现逐年递增趋势(见图1), 2014年达到4506篇, 预计2015年将突破5000篇。以上数据说明二代测序技术的相关研究正处于上升阶段, 更多的科研团队正投入到相关研究中。相比之下, 二代测序技术在法庭科学领域中的应用发展滞后, 截止2015年7月10日总共仅有119篇科研论文发表(见图2), 占比不足二代测序文章总数的1%, 且直到2014年文章数量才出现急剧增长, 说明二代测序技术在法庭科学领域起步晚, 尚处于刚刚兴起的阶段。
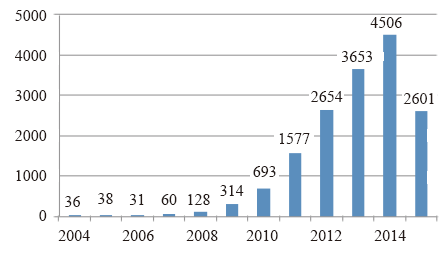 | 图 1 Pubmed历年新收录二代测序科研论文数量Fig.1 Annual statistics of research articles on NGS in Pubmed |
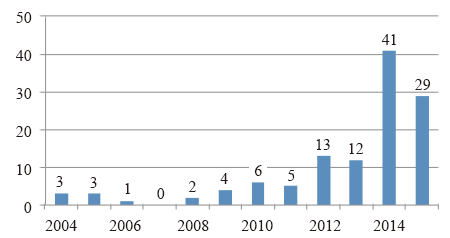 | 图 2 Pubmed历年新收录二代测序技术在法庭科学领域运用科研论文数量Fig.2 Annual statistics of research articles on forensic NGS in Pubmed |
STR技术是当前法医DNA领域的主流技术。首先, 我国公安机关现有400余个DNA实验室, 对于其中90%以上的实验室, 基于PCR-CE方法进行STR分析是唯一的技术手段; 第二, 目前国际上最先进的法医遗传学实验室仍视基于PCR-CE平台的STR分析为DNA鉴定金标准; 第三, 全球各国DNA数据库均基于STR基因座; 第四, 大量积案、冷案的DNA证据均以STR分型体现, 检材已无法再次获得。综合以上四点, 笔者认为STR技术在未来法医DNA领域中仍将占据统治地位, 二代测序技术若要在法医遗传学领域站稳脚跟, 则必须妥善解决好STR分型问题。
基于二代测序进行STR分析相比PCR-CE平台具有显著优势:第一, PCR-CE平台仅区分等位基因的片段大小, 核苷酸数量相等的所有等位基因被认为是同一个等位基因; 二代测序技术可明辨DNA序列, 展示出STR等位基因重复单元和侧翼序列的真实差异。丹麦哥本哈根大学Morling等利用二代测序技术对197份丹麦人样本进行STR检验, 在D12S391基因座中发现53种不同的等位基因, 而同批样本用PCR-CE平台检测, 仅发现15个不同的等位基因[14]。另一项研究显示, PCR-CE平台检测结果中30%的纯合子经测序验证为杂合子[13]。以上结果表明基于二代测序进行STR分析比PCR-CE平台更为精细化, 更充分发掘现有STR基因座的区分能力, 只需更少的基因座即可达到现有个体识别率。第二, 二代测序对降解检材进行STR分析具有优势。PCR-CE平台采用多色荧光复合扩增技术, 为在各色荧光中安置更多的基因座, 设计引物时经常有意保留较长的侧翼序列, 导致扩增效率降低, 不利于降解检材分型。利用二代测序技术进行STR分型, 各基因座片段长度完全可以重叠在小片段区域且互不干扰, 相当于将更多的常规STR基因座作为Mini-STR基因座使用, 对降解检材分型效果自然更好。
2011年, 丹麦自然历史博物馆Gilbert研究团队基于454平台率先将二代测序技术引入STR分析[15]。2012年, 比利时根特大学Deforce教授研究组比较了D3S1358等9个CODIS核心基因座在454和PCR-CE平台的测序效果[16]。Deforce教授肯定了利用454平台进行STR分析的可行性, 也客观指出454平台比PCR-CE平台更昂贵, 需要更繁重的实验室工作, 错误率也更高。同年, 美国巴特尔纪念研究Faith研究团队报道利用Illumina二代测序系统进行STR分型[17]。2014年, Morling等报道基于454平台进行STR分型尝试和方法优化, 研究了D3S1358、D12S391、D21S11等3个STR基因座, 发现30个新等位基因, 并在D12S391核心重复单元中发现新的SNP位点[14]。美国武装部队DNA鉴定实验室Scheible教授首次尝试利用二代测序进行案件检材的STR分型, 并建立了48个常用STR基因座的二代测序分型方法[18]。
2015年, Morling等利用Thermo Fisher公司的Ion PGMTM系统, 以及尚处于试用阶段的包含10个基因座的试剂盒产品, 建立了STR分型方法[19]。结果表明该方法在价格、需要检材量、测序速度等方面相比其它方法具有优势。目前, Ion PGMTM系统测序读长可达400 bp, 虽然该方法仅针对重复单元较短的基因座(103~205 bp), 但该系统仍具有潜力分型重复单元更长的基因座。Illumina公司推出的MiSeq FGxTM系统和Forenseq DNA Signature Prep试剂盒是现有唯一商业化二代测序STR分型解决方案, Illumina同时宣布要用二代测序技术替代PCR-CE平台。而Thermo Fisher公司的策略则是将二代测序STR分型作为PCR-CE平台的补充[13]。就在本文撰写过程中, 上海刑事科学技术研究院、上海市公安局、复旦大学联合研究团队报道在国际上首次成功利用二代测序技术进行Y-STR分型[20]。
不同于STR分型区分长度多态性, SNP分型区分的是序列多态性。在二代测序数据分析方面, SNP要比STR更简单。SNaPshot是基于PCR-CE平台的SNP分型方法, 也是目前最常用的SNP分型方法之一。澳大利亚Daniel研究组在136个SNP位点范围内比较了二代测序(Ion PGMTM平台)、SNaPshot和Sanger测序的实验结果, 二代测序分型成功率达97%以上, 也发现个别位点由于特殊结构而导致测序错误[21]。奥地利、西班牙、德国等国家实验室对二代测序SNP分型进行了实验室间比对研究, 研究组使用的是Ion PGMTM平台和配套试剂盒, 结果表明灵敏度达到25 pg, 仅在rs1979255等5个位点发现结果不一致情况[10]。美国国家标准与技术研究院Vallone团队研究了二代测序对降解DNA的分型效果, 发现基于二代测序Ion PGMTM平台的SNP体系对降解检材分型效果优于基于PCR-CE平台的STR、Mini-STR和Indel体系[22]。二代测序数据的增长对Y染色体进化和Y-SNP研究也产生了深远影响, 每次二代测序研究都有机会对Y染色体重新分析并发现新的Y-SNP, 学者已经呼吁在二代测序背景下尽快形成具有国际共识的系统命名规则[23]。
2014年以来, 法医遗传学相关二代测序研究最集中的领域当属线粒体基因组测序[24, 25, 26, 27, 28, 29, 30]。虽然线粒体DNA技术的应用远不及STR技术广泛, 我国公安机关开展线粒体DNA鉴定的实验室也相对较少, 但该技术对于涉及微量DNA检材或母系遗传调查的案件具有独特优势和不可替代性, 是一项重要的刑事技术手段[31]。线粒体基因组全长16569个碱基, 由于使用Sanger方法测线粒体基因组全序列费用高且工作量繁重, 很多法庭科学实验室只关注控制区中约600个碱基的高变区Ⅰ 和高变区Ⅱ 。二代测序技术使线粒体基因组测序的法医学常规应用成为可能, 线粒体DNA的识别率将获提升, 线粒体技术有可能重新焕发青春。
Irwin等人率先意识到现有法庭科学线粒体数据库只包含控制区信息, 无法满足线粒体基因组二代测序的数据分析需求[32]。Parson团队评估了Ion PGMTM系统对线粒体基因组进行测序的效果, 他们建立了64套线粒体基因组, 并全部与经典的Sanger测序结果比对, 发现结果差异率低于0.02% [11]。美国Budowle研究组和McElhoe研究组分别建立并优化评估了基于Illumina二代测序平台的线粒体基因组测序方法, 并比较了高变区Ⅰ /高变区Ⅱ 与线粒体基因组的单倍型多样性差异[25, 26]。与高变区序列分析类似, 线粒体基因组测序同样只需微量DNA样本。Parson等人使用Illumina二代测序平台成功地从单根毛干样本中恢复出完整的线粒体基因组序列, 并建立起法医DNA实验室可常规使用的技术方法[27]。454系统同样支持线粒体基因组测序[24, 29], 并被应用于异质性分析研究中[29]。
随着二代测序技术在法庭科学领域萌芽与发展, 一场革命性的技术变革正在酝酿之中。二代测序技术的优势并不局限于高通量、高速度、集成化、低成本, 在法庭科学领域, 二代测序技术最有可能从微量的生物检材中挖掘出案件所需要的全部遗传学相关信息, 这对于公安实战具有无可比拟的吸引力。二代测序技术在法庭科学领域的应用范围并不局限于本文提到的STR、SNP和线粒体基因组测序, 也可以为与法庭科学相关的动物、植物、微生物种属鉴定和来源分析提供解决方案, 服务于微生物恐怖袭击、濒危物种贩卖、食品安全等类型案件, 还可以用于表观遗传学和MicroRNA分析, 推断组织来源、嫌疑人年龄等信息[31]。
同时, 我们也清醒认识到二代测序距离法庭科学常规应用还有一定距离:首先, 测序成本必须有效降低。低成本是二代测序的显著优势, 也是各公司推广产品过程中的重要宣传项目, 但所谓“ 低成本” 是与二代测序的另一个显著特点“ 高通量” 相辅相成的, 即只有当测序数据量足够大时, 单位数据量的成本才能够降下来。当前, 常规STR分型、SNP分型往往只关注十几个到几十个位点, 在如此小的数据量前提下, 二代测序的成本则显著高于PCR-CE平台。第二, 法庭科学数据分析方法尚不成熟。以STR分型为例, PCR-CE平台关注的是STR的长度多态性, 而二代测序获得的是更加精细化的STR序列多态性, 将对法庭科学起到更大的支撑作用; 另一方面, 基于各国现有的庞大DNA数据库, 技术接轨是必然选择, 如何将序列多态性在一定程度上转化为长度多态性是二代测序工作者必须解决的问题。虽然业内公司已提出初步解决方案, 但大多是针对各个基因座的不同情况各个击破, 要实现统一的数据分析系统还需要更多的工作。第三, 数据准确性风险必须有效规避。法庭科学关乎个人命运、家庭幸福、公平正义和社会稳定, 是社会公众不允许出错的学科。各种二代测序平台都不可避免地存在一定比例的错误率, 这些技术风险的控制效果将决定二代测序技术何时可以通过法律认可应用于法庭科学实践。第四, 二代测序必须是一个开放的平台。一项技术要被普及运用, 离不开国际学界的广泛参与、探索、优化与完善。目前二代测序平台由少数公司提供, 除了价格、稳定性等因素外, 用户往往倾向于选择更加开放的平台, 即允许用户根据各自需求自主设计实验, 更改参数设置, 并与其它平台有效兼容。法庭科学新技术往往需要专家证人出庭作证, 只有整个技术的工作原理清晰明了, 才有可能被法庭和立法机关接受。丹麦Morling教授已经提出, 二代测序的数据分析软件算法必须公开, 数据分析算法的“ 黑盒子” 是无法被接受的[13]。第五, 数据整合与伦理学。如前文所述, 要充分发挥二代测序技术“ 高通量” 与“ 低成本” 的优势, 数据整合势在必行。基于PCR-CE平台开发的常染色体STR、Y染色体STR、地域种族推断SNP、外表特征刻画SNP、插入缺失多态性Indel、mRNA、表观遗传学修饰等多种实验体系, 在二代测序的技术框架下都可以整合在一起, 甚至法庭科学以外的其他标记(如疾病诊断、遗传缺陷筛查等)也可以整合在一起。从二代测序技术的特点角度讲, 越整合成本越低; 从伦理学的角度讲, 越整合伦理学风险越大; 如何把握好二者之间的平衡, 尚须全面深入的研究探讨。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|