
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
二乙酰吗啡盐酸盐标准物质的研制(2)—定值和不确定度评定
[冯超 , 白燕平]
, 白燕平]
, 白燕平]
|
|


作者简介:冯超(1981—),男,山东人,博士,主要从事毒品和易制毒化学品标准物质的制备和定值的研究工作。Tel:010-61957086; E-mail: fengchao@gab.ga
目的对研制的二乙酰吗啡盐酸盐标准物质进行定值,并评定定值结果的不确定度。方法采用定量核磁共振法和质量平衡法进行定值;采用液质联用法用于有机物杂质的定性分析,采用电感耦合等离子质谱法、离子色谱法、顶空-气质联用法和卡尔费休滴定法测定无机阳离子、阴离子、挥发性有机溶剂残留和水分等杂质的含量。结果定量核磁共振法的纯度为95.6%,不确定度为0.13%;质量平衡法的纯度为95.3%,不确定度为0.93%。结论二乙酰吗啡盐酸盐标准物质的纯度值为95.6%,扩展不确定度为1.2%(k=2)。
Objective To characterize the prepared diacetylmorphine hydrochloride reference material, and to evaluate the uncertainty of assessment of purity. Methods Diacetylmorphine hydrochloride was characterized by both quantitative-NMR (qNMR) and mass balance method. The organic impurities were identified by LC/MS-IT-TOF, and the contents of inorganic cations and anions, residual of volatile organic solvents and water in the reference material were assayed by ICP-MS, ion chromatography, Head-space GC-MS, Karl-Fischer titration methods, respectively. Results The qNMR purity was calculated to be 95.6% with standard uncertainty of 0.13%, while the mass-balance purity was 95.3% with standard uncertainty of 0.93%. Conclusion The certified purity value of diacetylmorphine hydrochloride was 95.6%, with the expanded uncertainty of 1.2% (k=2).
标准物质的定值是对标准物质特性量值赋值的全过程, 直接决定了标准物质量值的溯源性, 是标准物质研制过程中最重要和复杂的环节之一。根据标准物质的性质、潜在用途、定值实验室能力等因素, 可选择不同的方法模式进行标准物质的定值测量, 如基准测量方法模式、彼此独立的多个方法模式、实验室间比对研究等[1]。在化学成分纯度标准物质的研制中, 质量平衡法和定量核磁共振法(qNMR)是经常采用的定值分析测量方法。
质量平衡法定值的依据是, 待测标准物质主成分的含量与其中所含水分、残留溶剂、无机物(灰分)等杂质的含量之和应为100%。该方法的缺点是, 当样品中含有未知杂质时, 由于不同杂质响应值的差异, 可能无法准确确定杂质的含量[2]。定量核磁共振法用于纯度测定的基础是各化学环境中不同质子共振峰的面积只与所包含的质子数有关, 不需引进任何校正因子就可直接根据各共振峰的积分值推算所代表的自旋核的数量, 具有快速、简便、准确、专属性高、无需使用纯品参比物等特点[3]。本文使用质量平衡法和定量核磁共振法两种方法对研制的二乙酰吗啡盐酸盐标准物质[4]进行了定值。
1.1.1 仪器设备 AVANCE III型核磁共振谱仪(瑞士布鲁克公司), 400 MHz。
高效液相色谱仪1200型, 二极管阵列检测器(DAD), (美国安捷伦公司)。
液相色谱串联质谱仪LCMS-IT-TOF(日本岛津公司)。
单四极杆气相色谱质谱联用仪ISQ型, 配备TRACE GC ULTRA气相色谱仪、TriPlus RSH顶空进样器(美国热电公司)。
电感耦合等离子体质谱仪7700型(美国安捷伦公司)。
离子色谱仪ICS 5000型(美国戴安公司)。
卡尔-费休水分仪852 Titrando型(瑞士万通公司)。
分析天平XP6型(美国梅特勒-托利多公司)。
1.1.2 化学试剂 苯甲酸(美国国家标准与技术研究院, SRM 350 b), 标准值99.9978%± 0.0044%(k=2)。
O6-单乙酰吗啡(中国食品药品检定研究院)。氘代甲醇(美国Cambridge Isotope Laboratories公司)。正己烷、氯仿、丙酮、乙醚(色谱纯, 美国J.T.Baker公司); 乙酸乙酯(HPLC级, 美国Burdick & Jackson公司); 异丙醇(HPLC级, 美国MREDA公司); 二氯甲烷(HPLC级, Cleman Chemical公司); 乙腈、乙醇(色谱纯, 美国Fisher公司); N, N-二甲基甲酰胺(DMF)(百灵威科技有限公司)。
元素混合标准溶液(10 mg/L ): Li、Be、Na、Mg、Al、K、Ca、V、Cr、Mn、Fe、Co、Ni、Cu、Zn、Ga、As、Se、Rb、Sr、Ag、Cd、Cs、Ba、Tl、Pb、U(美国安捷伦公司8500-6940);
F-溶液标准物质GBW(E)080549; Cl-溶液标准物质GBW(E)- 080268; S
1.2.1 质量平衡法 (1)高效液相色谱(HPLC)分析条件。色谱柱Phenomenex Luna C18(2), 250mm× 4.6mm, 5μ m; 流动相30mmol/L NH4Ac水溶液∶ 乙腈=60∶ 40; 流速1ml/min; 检测波长285nm。
(2)无机阴离子杂质分析条件。色谱柱IonPac AS11, 4mm× 250mm, 4μ m; 淋洗液:氢氧化钾, 30mmol/L; 流速1.5mL/min; 抑制器ASRS 4mm; 检测器:电导检测器;
分别绘制F-、Cl-、Br-、N
(3)无机阳离子杂质分析条件。分析方法采用电感耦合等离子体质谱(ICP-MS)半定量法; 样品准备:准确称取样品15mg, 溶于2mL 5%的稀硝酸中。
测定元素:Li, Na, Mg, Al, K, Ca, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, As, Se, Br, Kr, Rb, Sr, Y, Zr, Nb, Mo, Ru, Rh, Pb, Ag, Cd, In, Sn, Sb, Te, I, Cs, Ba, La, Ce, Pr, Nd, Gd, Dy, Ho, Tm, Ta, Os, Au, Hg, Pb, Bi共54种阳离子。
(4)挥发性有机溶剂残留(VOC)分析条件。顶空条件:顶空瓶加热温度80℃, 进样温度90℃, 传输线温度90℃; 样品平衡时间30min; 进样体积1mL;
色谱条件:色谱柱VF-624ms, 30m× 0.25mm I.D.× 1.40μ m; 载气为氦气, 流速为1.0ml/min; 进样口温度200℃; 分流比10/1, 分流流速10ml/min; 升温程序:32℃(2.5min), 2℃/min至45℃, 30℃/min至180℃(0.5min);
质谱条件:电子轰击离子源, 碰撞能量70eV; 离子源温度230℃; 传输线温度280℃; 扫描范围m/z 20~150, 驻留时间0.2s;
测定乙醇、乙醚、丙酮、异丙醇、二氯甲烷、正己烷、乙酸乙酯、氯仿等8种有机溶剂混合标准溶液和二乙酰吗啡盐酸盐标准物质样品中残留溶剂的GC/MS TIC谱图、8种有机溶剂定量离子的GC/MS SIM谱图和二乙酰吗啡盐酸盐标准物质样品中残留溶剂定量离子的GC/MS SIM谱图, 并绘制8种有机溶剂的标准曲线。
1.2.2 定量核磁共振法 准确称量二乙酰吗啡盐酸盐样品和苯甲酸标准物质各约10mg, 加入约1mL氘代甲醇溶解; 探头1H-13C 5mm Z向梯度反向观察探头;
实验参数:90° 脉冲, 扫描范围-0.5ppm~8.5ppm, 采集时间10s, 延迟时间50s, 扫描次数16次, 实验温度300K, 积分范围约200Hz。
2.1.1 水分杂质定量 经测定, 二乙酰吗啡盐酸盐标准物质中水分的质量百分含量为3.23%(见表1)。
| 表1 二乙酰吗啡盐酸盐标准物质所含水分的测定结果 |
水分测量的不确定度计算公式如下:
urel (Xw )=
其中, urep是测量重复性产生的不确定度, u(m)是样品称量产生的不确定度, u(m’ )是样品中绝对水量测量产生的不确定度, u(a1)是碘的生成反应电解效率的不确定度, u(a2)是副反应造成碘和水的化学计量比例偏离1∶ 1产生的不确定度, u(aa)是未释放水量产生的不确定度。将上述6部分不确定分量按照公式1合成, 得到合成相对标准不确定度urel(Xw)=0.20。因此, 水分测量的标准不确定度为:
u(Xw)=urel(Xw)× Xw=0.20× 0.0323=0.0065。
2.1.2 无机阳离子杂质定量 经ICP-MS半定量法测定, 二乙酰吗啡盐酸盐标准物质中无机阳离子总的质量百分含量为0.223%。
根据文献[5], ICP-MS半定量法测定元素含量的相对不确定度为0.3。因此, 无机阳离子测定的标准不确定度为:
u(Xm)=urel(Xm)× Xm=0.3× 0.22%=0.00066。
2.1.3 无机阴离子杂质定量 如表2所示, 二乙酰吗啡盐酸盐标准物质中含有大量的氯离子, 证实其确以盐酸盐形式存在。除氯离子外的阴离子主要是氟离子, 其百分含量为0.47%。
| 表2 二乙酰吗啡盐酸盐标准物质无机阴离子的测定结果 |
无机阴离子杂质含量Xa的计算公式为:
其中, V为溶解样品的水的体积, m为样品质量, c1至c5分别为样品中氟、溴、硝酸根、硫酸根、磷酸根等阴离子杂质的浓度。因此, 无机阴离子含量测定的不确定度为:
式中, urep是测量重复性产生的不确定度, u(V)是溶解样品的水的体积产生的不确定度, u(m)是样品称量产生的不确定度, u(ci)是各种阴离子杂质的校准用标准溶液配制产生的不确定度, u(cs)是由校准曲线求得的样品中某种阴离子杂质的浓度产生的不确定度。将上述5部分不确定度分量按照公式3合成, 得到合成相对标准不确定度urel(Xa)=0.040, 因此, 无机阴离子杂质测定的标准不确定度为:
u(Xa)=urel(Xa)× Xa=0.040× 0.47%=0.00019。
2.1.4 挥发性有机溶剂残留定量 经测定, 二乙酰吗啡盐酸盐标准物质中残留较多的有机溶剂是异丙醇和丙酮, 挥发性有机溶剂残留的总量为0.38%(见表4)。
| 表4 二乙酰吗啡盐酸盐标准物质挥发性有机溶剂的测定结果 |
挥发性有机溶剂残留含量Xv的计算公式为:
其中, mDMF为溶解样品的DMF的质量, ci为样品中某种残留有机溶剂的浓度, m为样品的质量。因此, 挥发性有机溶剂残留含量测定的不确定度:
其中, urep是测量重复性产生的A类不确定度, u(mDMF)是溶解样品的DMF的质量称量产生的不确定度, u(m)是样品称量产生的不确定度, u(ci)是各种挥发性有机溶剂杂质的校准用标准溶液配制产生的不确定度, u(cs)是由校准曲线计算样品中某种挥发性有机溶剂残留杂质的浓度产生的不确定度。将上述5部分不确定度分量按照公式5合成, 得到合成相对标准不确定度urel(Xv)=0.51, 因此挥发性有机溶剂残留测定的标准不确定度为:
u(Xv)=urel(Xa)× Xv=0.51× 0.38%=0.0027
2.1.5 有机物杂质分析 如表5所示, 二乙酰吗啡盐酸盐标准物质的HPLC面积归一化法的纯度为99.6%。
| 表5 HPLC面积归一化法测定结果 |
HPLC法测定纯度的不确定度计算公式为:
urel
其中, 不确定度的4个分量分别为液相色谱法定值测量重复性引入的不确定度、各组分在不同检测波长下响应差异引入的不确定度、仪器检测线性引入的不确定度和仪器检测限(LOD)引入的不确定度。经计算, 合成相对标准不确定度urel(P0)=0.0066。
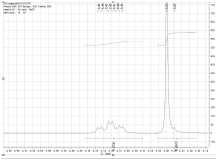
如图1所示, 采用LC/MS-IT-TOF测定出二乙酰吗啡盐酸盐标准物质中保留时间为4.12min的杂质, 其质荷比为328.1525, 相对分子质量为327, 结合其碎片离子可以推断该杂质为O6/O3-单乙酰吗啡。
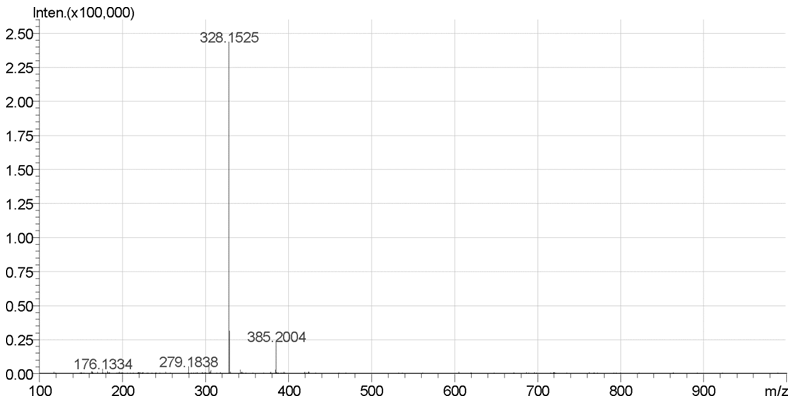 | 图1 二乙酰吗啡盐酸盐标准物质样品中Rt=4.12min的杂质的质谱图 |
2.1.6 定值结果 质量平衡法计算标准物质纯度的公式为:
其中, PHPLC为高效液相色谱纯度、Xv为挥发性有机溶剂含量、Xv为水分百分含量、Xv为无机阳离子含量、Xa为无机阴离子含量。根据前文中所述测定结果和不确定度评定结果, 计算得到二乙酰吗啡盐酸盐标准物质的质量平衡法纯度为95.3%, 其测定的标准不确定度为:
2.2.1 定量峰的选择 对二乙酰吗啡盐酸盐标准物质和苯甲酸标准物质分别进行1H-NMR分析, 从两者的1H-NMR谱图中互不重叠且没有杂质峰干扰的的谱峰作为定量峰。
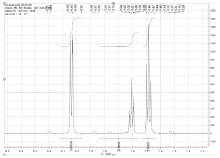
如图2所示, 苯甲酸的1H-NMR谱图中, 化学位移7.59ppm~7.62ppm和7.46ppm~7.50ppm的谱峰都有杂峰干扰; 只有化学位移8.03ppm~8.05ppm的谱峰无干扰, 可以用作内标物苯甲酸的定量峰。
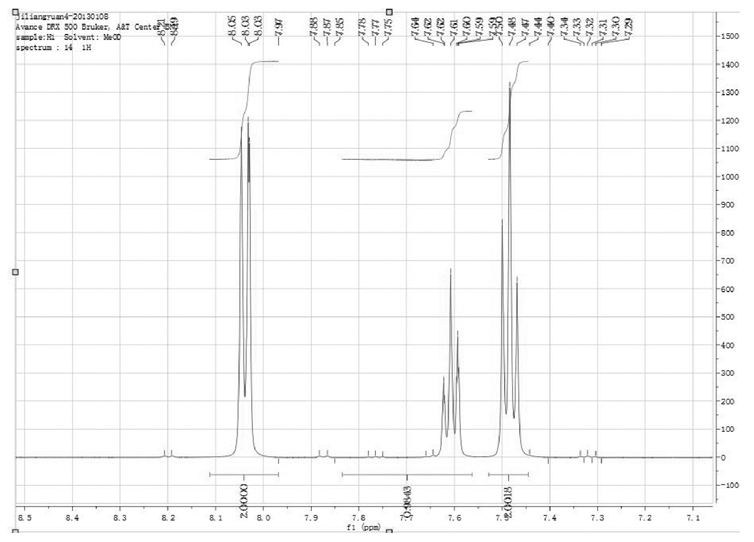 | 图2 苯甲酸的1H-NMR谱图 |
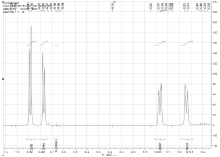
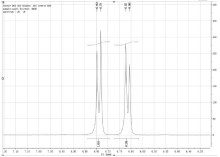
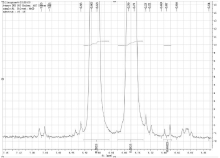
根据理论化学位移值推算, 二乙酰吗啡盐酸盐与杂质O3/O6-单乙酰吗啡化学位移相比, 只有二乙酰吗啡盐酸盐1位[2.28(3H, s)ppm]、6位[5.53-5.55(1H, d)ppm]和9位[6.88-6.90(1H, d)ppm]上H的化学位移值与O6-单乙酰吗啡不同, 可以用作定量峰。通过分析二乙酰吗啡盐酸盐的1H-NMR谱图, 化学位移2.28ppm的谱峰的右肩上存在小杂质峰(见图3), 会导致测定结果偏高, 因此不能选为二乙酰吗啡盐酸盐的定量峰; 而化学位移5.53ppm~5.55ppm和6.88ppm~6.90ppm的谱峰则可以选作定量峰(见图4)。
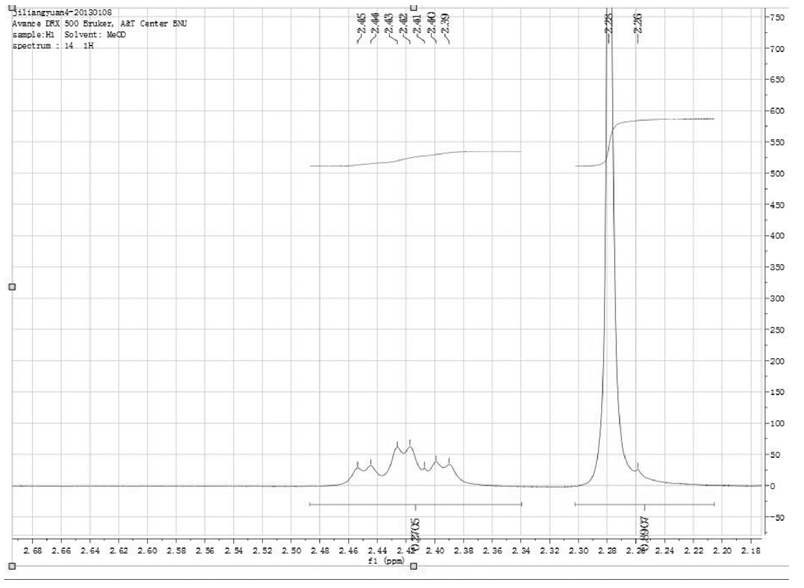 | 图3 二乙酰吗啡盐酸盐标准物质化学位移2.28ppm的共振峰 |
 | 图4 二乙酰吗啡盐酸盐化学位移6.88ppm~6.90ppm和5.53ppm~5.55ppm的共振峰 |
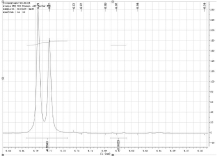
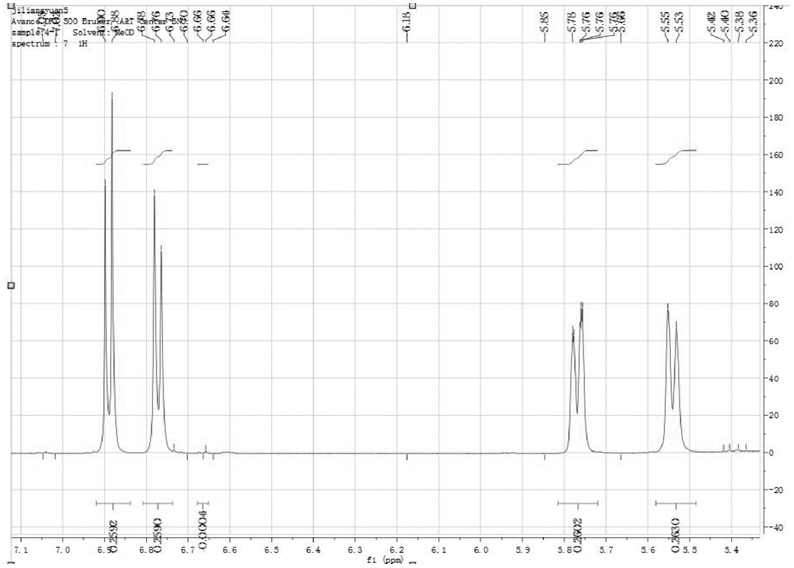
其他化学位移处的谱峰如果实现了基线分离, 通过减去杂质O3/O6-单乙酰吗啡的干扰, 也可以用于定量, 例如化学位移6.77ppm~6.79ppm的谱峰(见图5)。
 | 图5 二乙酰吗啡1H-NMR谱图中化学位移6.67ppm~6.69ppm的谱峰 |
如图5所示, 化学位移6.67ppm~6.69ppm的谱峰可能是杂质O3/O6-单乙酰吗啡的峰。为此, 测定O6-单乙酰吗啡标准物质的1H-NMR谱图, 并观察到化学位移6.66ppm~6.67ppm和6.78ppm~6.80ppm的谱峰(见图6)。因此, 二乙酰吗啡盐酸盐1H-NMR谱图中化学位移6.67ppm~6.69ppm的谱峰, 确实为O6-单乙酰吗啡的共振峰。
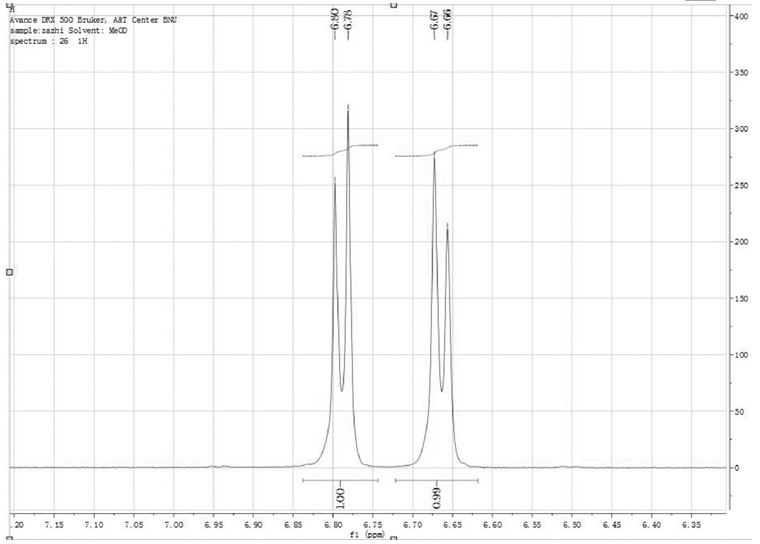 | 图6 O6-单乙酰吗啡的6.66ppm~6.67ppm和6.78ppm~6.80ppm的谱峰 |
从图6中还可以看出, O6-单乙酰吗啡化学位移6.78ppm~6.80ppm的谱峰与二乙酰吗啡盐酸盐化学位移6.77ppm~6.79ppm的谱峰重叠, 后者用作定量峰, 就要通过O6-单乙酰吗啡6.67ppm~6.69ppm的谱峰计算出O6-单乙酰吗啡的量后, 进行扣除(见图7)。
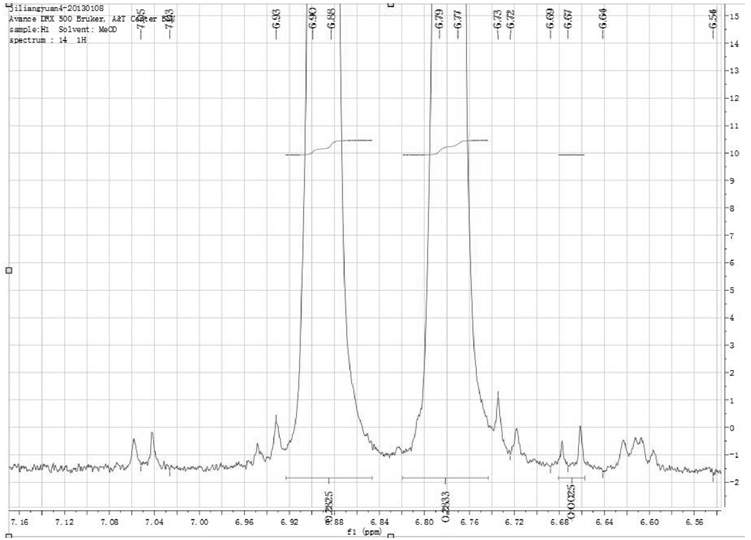 | 图7 二乙酰吗啡盐酸盐谱图中杂质O6-单乙酰吗啡6.67ppm~6.69ppm的谱峰 |
2.2.2 定量计算结果 qNMR法纯度的计算公式如下:
其中, Px为待测物的纯度、Ix为样品定量峰的积分面积、Istd为内标物定量峰的积分面积、Nstd为内标物定量峰的核群核个数、Nx为样品定量峰的核群核个数、Mx为样品的分子量、Mstd为内标物的分子量、mstd为添加的内标物的质量、m为样品的称样量、Pstd为内标物的纯度。
综合以上定量峰的选择, 以某次测定为例, qNMR法对二乙酰吗啡盐酸盐标准物质的定量结果如表6所示。
| 表6 定量核磁共振法定量峰的选择结果 |
平行测定6个样品, 取平均值, 得到二乙酰吗啡盐酸盐标准物质的qNMR法定值结果为95.6%。
2.2.3 不确定度评定 根据公式9, 定量核磁法定值的不确定度评定公式为:
式中各项依次表示重复性引入的不确定度、分子量的不确定度、称量引入的不确定度和内标物纯度的不确定度。因此, qNMR法定值结果的标准不确定度uqNMR=0.0013。
采用F检验比较上述两种定值方法是否等精度, 统计量F的计算公式为:
其中, s1和s2分别为两种定值方法的标准偏差。
F检验要求:
fα (ν 1, ν 2) < F < 1/[fα (ν 1, ν 2)] (公式17)
其中, ν 1和ν 2分别为两种定值方法的自由度; 置信度α 取95%, 由定值结果计算得到F=11.1; 经查表, f0.95(5, 5)=5.05, 1/[ f0.95(5, 5)]=0.20, 不满足公式17。因此, 质量平衡法和定量核磁共振法这两种定值方法不等精度。但是由于两种定值方法的结果相近, 可采用加权平均法计算标准物质纯度值y:
其中, 权重
根据ISO Guide 35[6], 标准物质纯度值的不确定度包括三个部分:两种不同方法定值的不确定度; 标准物质瓶间不均匀性产生的不确定度; 标准物质特性量值的长期和短期稳定性分别引起的不确定度。
(1)两种不同方法定值的不确定度。因为质量平衡法和定量核磁共振法的定值不等精度, 所以两种不同方法定值的不确定度计算公式为:
(2)标准物质的瓶间不均匀性引入的不确定度
(3)标准物质的长期稳定性引入的不确定度
在95%置信概率下取包含因子
所以, 二乙酰吗啡盐酸盐标准物质的定值结果为:
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|