



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
大量环己胺中甲基苯丙胺的定性分析
引用本文
黄星, 王蔚昕, 张春水. 大量环己胺中甲基苯丙胺的定性分析[J]. 刑事技术,2012,37(6): 55-57 
Permissions
Copyright©2012, 《刑事技术》编辑部
《刑事技术》编辑部
大量环己胺中甲基苯丙胺的定性分析
关键词:
甲基苯丙胺; 环己胺; 气质联用
中图分类号:DF795.1
文献标志码:A
文章编号:1008-3650(2012)06-0055-03
甲基苯丙胺是新型毒品中最常见的一种。毒贩为谋取更多利益、降低交易风险, 在甲基苯丙胺晶体中掺入与甲基苯丙胺晶体外观极为相似的物质[1, 2, 3]。笔者所在实验室, 近期对几例疑似冰毒案件进行检验, 发现所送检材中均使用环己胺作为掺假剂, 且掺假量极大, 对这些检材使用常规方法进行检验, 掺入的大量环己胺对甲基苯丙胺的定性分析产生严重干扰。针对此问题, 通过对方法的优化, 实现了甲基苯丙胺、环己胺的定性分析, 与常规方法比较可得到更满意的结果。
1 实 验
仪器和试剂:Agilent 7890N毛细管气相色谱仪, 配置5975C MS。XS-105电子天平购自梅特勒公司。甲醇、甲苯, 分析纯(北京化工厂)。
气相色谱条件:色谱柱HP-5MS, 30m × 0.25mm × 0.25μ m; 分流比为20:1;
升温速率:60℃(0min) 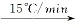
质谱参数:电离源EI, 电离电压70eV, 倍增器电压1.0kV, 扫描质量范围40~500amu, 全扫描方式, 溶剂延迟时间2.5min, 其它条件为调谐值。
取送检的检材, 用玛瑙研钵将其研磨成均匀细粉后称取适量, 装入试管中, 加入甲醇或甲苯适量, 振荡10min, 静置, 取1.5mL上清液装入自动进样器小瓶中, 供GC-MS分析。
2 结果与讨论
2.1 检材经甲醇溶解后GC-MS分析结果
对环己胺盐酸盐及甲基苯丙胺标准样品的甲醇溶液进行GC-MS分析, 发现环己胺及甲基苯丙胺的出峰位置分别在2.621min和5.463min。
称取案件检材10mg, 用20mL甲醇溶解(样品浓度为0.5mg· mL-1)后供GC-MS分析(见图1a、1b), 2.621min处有明显出峰, 进行谱库检索, 证明出峰位置及离子碎片与标准物质高度匹配(见图2), 证明该组分为环己胺成分, 但未检出甲基苯丙胺成分。为防止漏检, 增加检材用量至100mg, 并定容在10mL甲醇中(样品浓度为10.0mg· mL-1), 上清液供GC-MS分析, 分析结果见图3显示, 由于环己胺浓度过大导致过载, 环己胺的拖尾峰已延伸到5.371min之后, 严重影响了甲基苯丙胺的检测。
 | 图1a 检材经甲醇溶解后GC-MS检验的TIC, 样品浓度为0.5mg· mL-1 |
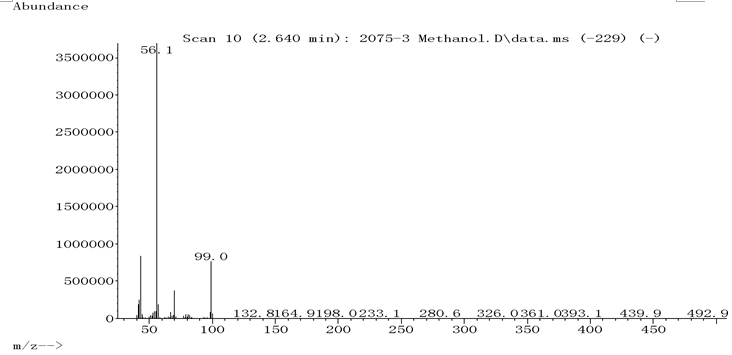 | 图1b 2.621min对应组分的离子碎片图谱 |
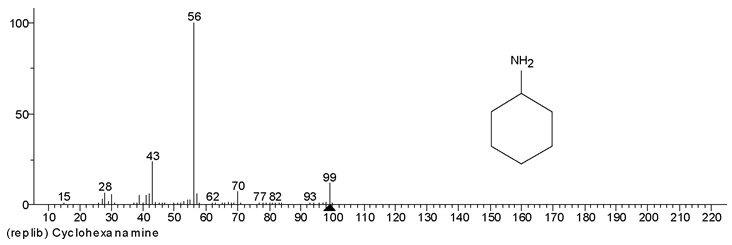 | 图2 环己胺谱库检索结果 |
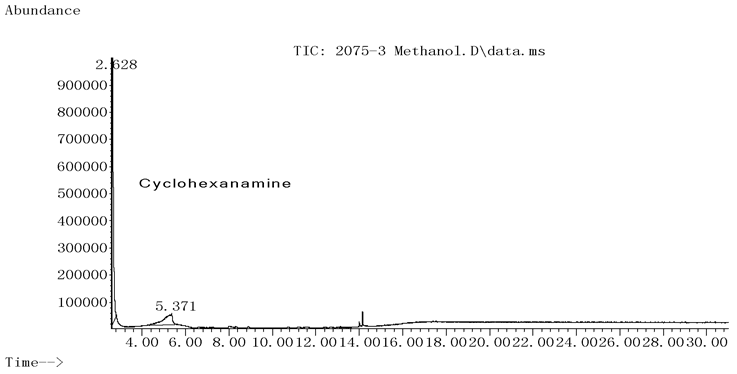 | 图3 检材经甲醇溶解后GC-MS检验的TIC, 样品浓度为10.0mg· mL-1 |
2.2 优化方法下检材GC-MS分析结果
经过实验发现, 环己胺在水、酸液、碱液、甲醇、乙醇、氯仿、乙酸乙酯中都具有很好的溶解性。通过比较环己胺(见图2)和甲基苯丙胺(见图4)的结构式, 基于甲基苯丙胺含有苯环这一结构特征, 并结合相似相容原理, 欲采用甲苯作为溶剂从大量环己胺中提取可能存在的微量甲基苯丙胺。
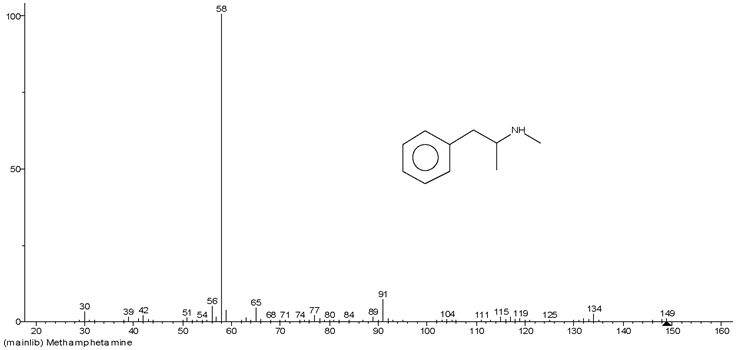 | 图4 甲基苯丙胺谱库检索结果及结构式 |
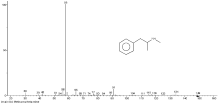
首先进行了溶解度实验, 将一定量环己胺溶于甲醇, 分别配制0.001、0.005、0.01、0.1、1.0、2.0、10.0mg/mL的环己胺甲醇溶液, 肉眼观察在各配制浓度下环己胺均完全溶解于甲醇, 在相同的检测条件下, 对各浓度环己胺甲醇溶液进行分析, 环己胺的检测灵敏度可达到0.005mg/mL, 而10.0mg/mL的环己胺甲醇溶液样品无法在现有检测条件下形成尖锐的色谱峰, 且环己胺峰宽大于3min, 不能用于环己胺的定性定量检验。进而, 将一定量环己胺溶于甲苯, 分别配制0.005、0.01、0.1、1.0、2.0、10.0mg/mL的环己胺甲苯溶液, 肉眼观察在各配制浓度下溶液中均有不溶解的晶体, 离心后, 对上清液进行GC-FID分析, 结果显示, 对各溶液均未检出环己胺, 可见环己胺在甲苯中的溶解度低于0.005mg/mL。由此可见, 环己胺在甲醇和甲苯中的溶解度有明显差异, 称取100mg所送案件检材, 并定容在10mL甲苯中, 发现与“ 2.1” 中的实验现象有明显不同:“ 2.1” 中使用甲醇作为溶剂, 检材能够全部溶解, 但是同样称样量和定容体积下, 以甲苯做溶剂, 有大量的晶体不能溶解。离心后, 提取甲苯体系的上清液供GC-MS分析, 分析结果见图5显示:在TIC中2.620min附近未检出环己胺成分, 在5.479min处检出甲基苯丙胺成分。经过GC-MS的定量分析, 该方法下检出所送案件检材中甲基苯丙胺的含量约为0.1%。
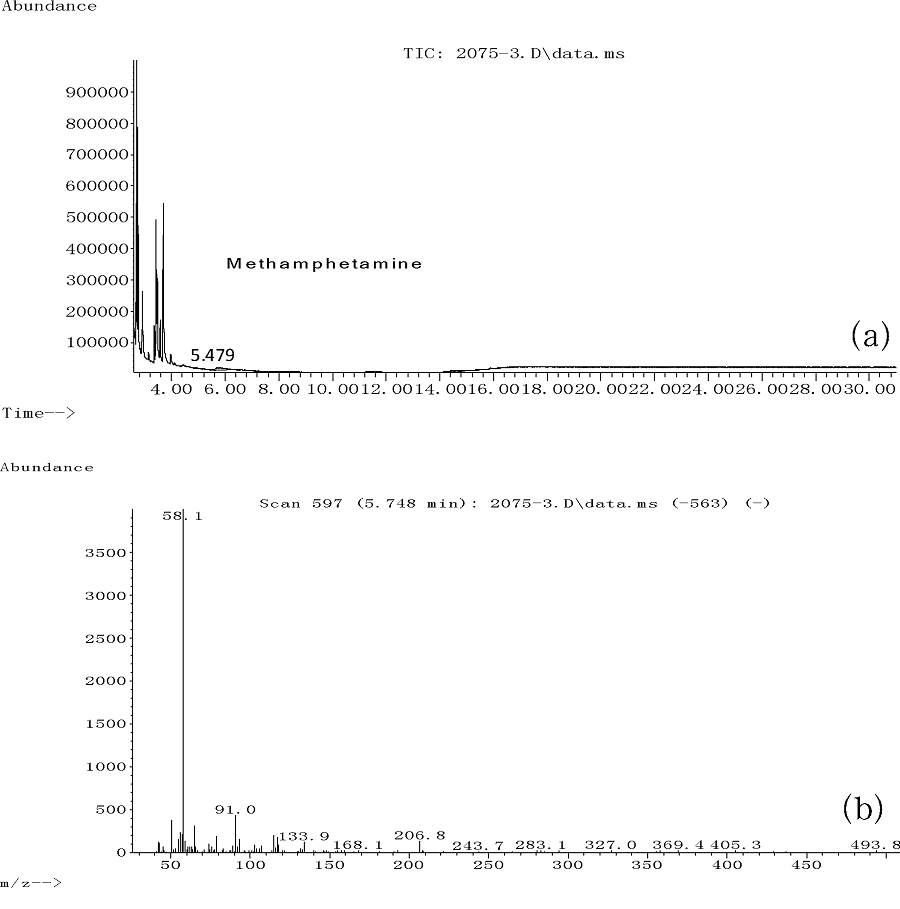 | 图5 以甲苯为溶剂检材提取液GC-MS分析的TIC(a)和检出甲基苯丙胺成分的质谱图(b) |
3 讨 论
本文采用甲苯为提取溶剂, 以检测大量环己胺中的微量甲基苯丙胺成分。该方法显示, 基于溶解度的不同, 排除了在甲醇溶解体系中大量环己胺对微量甲基苯丙胺(甲基苯丙胺浓度低至约为0.1%)定性分析的严重干扰, 可防止因方法不适当导致的漏检。
The authors have declared that no competing interests exist.