
{kind=link}
混合样本拆分查询犯罪嫌疑人的应用研究
[苑美青 , 李万水, 康艳荣, 刘冰, 彭建雄, 李盛, 谢群, 赵钊]
, 李万水, 康艳荣, 刘冰, 彭建雄, 李盛, 谢群, 赵钊]
, 李万水, 康艳荣, 刘冰, 彭建雄, 李盛, 谢群, 赵钊]
|
|


作者简介:苑美青(1977— ),女,主检法医师,博士,主要从事法医遗传学检案和研究工作。E-mail:yuanmeiqing000@yahoo.com.cn
目的 对一起杀人案中的混合样本进行拆分,期望获得嫌疑人的单一分型,以便于DNA数据库检索查询。方法 采用专家人工拆分和图挖掘拆分对混合样本进行拆分,结果入“DNA快速协查比对平台”检索。结果 2种方法先后比中数据库中同一前科人员。结论 对2人份混合样本进行拆分,在DNA数据库中直接查找犯罪嫌疑人,是一种新的办案思路。
Objective To obtain a single genotype of a suspect to facilitate database search, the DNA mixtures in a homicide case were de-convolved.Methods Mixtures component de-convolution using graph theory and expert analysis methods were used to analyze the mixture, the deduced genotypes were searched in the National DNA database.Results The same convicted was matched by either method.Conclusion DNA mixtures de-convolution followed by DNA Database search is a new approach in casework.
在法医DNA检验中, 经常会遇到混合样本的问题, 其中2人份混合样本所占比例最大。能为案件侦破提供线索和证据的2人份混合样本类型包括:受害人与嫌疑人样本混合, 2个嫌疑人样本的混合, 或者是嫌疑人与另一无关个体的混合样本。在这3种混合类型中, 第一种情形最为常见, 研究案情时该类检材与案件的关联度较高, 因而也最具分析价值。
近年来, 国外在混合样本的拆分分析方面, 报道了一些有参考价值的方法和案例, 特别是在没有对照样本的情形下, 如何在数据库中直接检索查询犯罪嫌疑人, 但目前国内尚未见此类报道。本文对一起案件中的 2人份混合样本进行了专家人工拆分和图挖掘拆分, 分别推断出嫌疑人可能具有的分型, 再通过“ DNA快速协查比对平台” 检索, 均直接比中同一前科人员, 使得案件在4天内快速侦破。
在对样本分析之前, 首先应确定样本是否为混合样本, Lafountain[1]等认为, 混合样本的判断标准有三个:(1)基因座出现的等位基因个数, 超过两个就提示可能存在混合(除了极个别位点的三倍型); (2)杂合子两个等位基因峰高不平衡, 标准为杂合子中2个等位基因峰高比小于60%; (3)stutter带的峰高大于15%。如果这三项都符合的话, 就可以确定该样本为混合样本。
1.2.1 钟摆查询系统[5](Pendulum Search) 该系统是利用最小二乘法(least squares method)估计2人份混合样本的混合比例, 然后按照杂合子平衡原则计算并排列出所有可能的基因型组合, 并按照可能性大小降序排列。此方法由英国法庭科学服务部(Forensic Science Service, FSS)提出, 并开发出商业化的自动分析软件(FSS-i3中的i-Stream模块), 输出结果可导入数据库中进行批量检索查询。
1.2.2 图挖掘拆分(Mixtures Component Deconvolution Using Graph Theroy, MDG) 国内康艳荣等[6]提出以图的挖掘算法为基础的MDG算法, 对混合STR 图谱中每个基因座“ 构造” 出不同的等位基因图, 形成等位基因图集, 把DNA混合样本的拆分转化为在图集中的挖掘问题, 最后得出混合样本的所有可能的拆分组合。目前已开发出较为成熟的软件系统, 相比钟摆系统拆分出完全正确分型的几率更高, 排名也更靠前, 但目前还无法批量导入数据库中进行比对, 需要手工录入。
1.2.3 专家人工拆分 有时, 当有一个已知样本的DNA分型时, 有经验的专家也会根据STR图谱对2人份混合样本进行人工拆分, 主要依据是所有基因座上2个贡献者的比例应保持相对恒定。首先通过1个4条带的基因座推断出大致的混合比例, 然后以此为根据“ 扣除” 掉已知样本的贡献部分, 既可拆分出另外一个未知样本的可能分型。
在没有已知样本做参考时, 如果2个组份的比例相差较大, 如1:3以上, 也可尝试着进行人工拆分。首先通过4条带的基因座等位基因峰高比例计算大致的混合比例, 然后推断主要贡献者和次要贡献者的排列组合方式。在拆分3条带、2条带的基因座时可能存在一定困难, 应进行不断的试错(trial-and-error), 最后得出多个可能的拆分组合, 结果类似钟摆查询或图挖掘拆分, 但数量会少得多。
某年9月15日, 黑龙江某市中年妇女刘某在等待接孩子放学的学校门口被人杀害, 受害人未遭明显的性侵害, 现场勘验分析可能有猥亵、搏斗等身体接触。办案单位除提取死者阴道拭子外, 还重点提取了死者指甲、面颊拭子、颈部扼压痕拭子、死者衣物等。
用两步法提取死者阴道拭子, 用M48法提取其余检材DNA, 以ID PLUS进行扩增、ID-X对电泳结果进行分析。结果死者阴道拭子上未获得STR分型; 死者指甲、面颊拭子、颈部扼压痕拭子及死者衣物上检出混合STR分型, 其中除包含死者本人的分型外, 还包含一男性个体的分型, 但不是死者丈夫所留, 分析应为罪犯所留。因没有嫌疑人可供比对, 对死者指甲的混合图谱进行拆分后, 录入DNA数据库检索查询嫌疑人。
将混合STR图谱进行专家人工拆分, 推断出未知男性个体12个基因座上的STR分型, 另外3个基因座难以拆分。将12个基因座的分型手工录入“ DNA快速协查比对平台” , 进行无容差模式下的检索查询。
同时, 在ID-X中导出混合DNA分型数据, 再导入公安部物证鉴定中心开发的MDG软件进行图形挖掘拆分。对前500对拆分结果进行分析, 并将第一对组合手工录入“ DNA快速协查比对平台” , 进行容差模式下的检索查询。
专家人工拆分的结果见表1。将表1中的基因型录入“ DNA快速协查比对平台” , 比中犯罪嫌疑人王某, 1天后将王某抓捕归案, 王某对犯罪事实供认不讳, 案件成功告破。
同时将混合图谱在ID-X导出后经图挖掘拆分后, 取前500对结果。将拆分的第1对基因型手工录入国家库进行快速比对, 在容差2对半条件下, 也比中犯罪嫌疑人王某。对拆分出的前500对组合进行分析, 12个及以上基因座完全比中的个数为47个, 具体结果见表2。
| 表1 受害人、混合样本、嫌疑人及人工拆分STR分型结果 |
| 表2 3号混合样本图挖掘拆分的前500对基因型与犯罪嫌疑人基因型匹配度 |
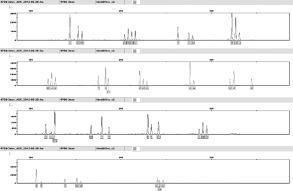 | 图1 死者指甲内容物获得的混合STR峰谱 |
混合样本的拆分一般都基于以下两种假设:(1)混合样本在同一基因座上的多个等位基因是共同扩增的; (2)样品的原始混合比例经过PCR扩增后也应保持不变, 所以普遍认为等位基因的峰高(峰面积)与原始浓度是成正比的。
混合样本拆分较难的原因多种多样:主要贡献者(major contributor)和次要贡献者(minor contributor)不明显, 某些基因座等位基因降解, 扩增产生的杂合子不平衡, 不同基因座之间DNA质量比例发生变异等等[7]。由于各种原因的限制, 即使是专家系统辅以精确计算的软件分析也不能保证预测到所有的随机事件。
对于混合样本的拆分方法, 不同的研究者提出了不同的方法, 国外关于DNA混合样本拆分问题研究始于上世纪90年代, 主流的拆分方法主要是频率学派和贝叶斯学派各自提出的方法, 如频率学派提出的PE法[8], 贝叶斯学派提出的用于定性数据分析的定性分析法[9], Gill[10]等人则采用分析峰面积的残余量, 排除一些不大可能的基因型组合。Perlin 等[11]提出了线性混合样本分析方法, Martin[5]等人提出的钟摆系统, Wang[7]等提出最小二乘法拆分, 此3人不同程度的利用了电泳中峰高/峰面积的定量信息, 利用最小二乘法原理寻找最可能的混合样本中的基因型。康艳荣等提出以图的挖掘算法为基础的MDG算法, 最后得出混合样本的所有可能的拆分组合。系统研究显示, 该软件在前100、200、500对中获得完全正确的分型组合的概率分别为75%、87%、93%, 目前在我国DNA数据库无法批量进行数据比对的情况下, 本文提出的将第1对分型手工录入数据库中, 进行容差模式下的检索查询, 不失为一种方便可行的办法。
一般来说, 软件拆分比人工拆分保守, 有时因为扩增不平衡等原因使得某个等位基因的峰面积过低时, 软件拆分可能得不到正确的分型, 有统计表明, 专家人工拆分与钟摆系统软件拆分没有区别的占37%, 人工拆分比钟摆系统用到更多排列组合的占5% , 钟摆系统拆分出更多排列组合的占49%[5]。在实际应用中, “ Pendulum” 算法中, 混合样本中主要贡献者的分型被正确拆分出来且排在第一位的可能性有50%, 主要贡献者或次要贡献者分型均正确拆分且排在前10名的可能性有40%[5]。
专家人工拆分因为要凭经验和判断, 难免具有主观性。Clayton[2]等人强调混合样本分析的独立性, 即单独分析混合样本的基因型组合, 最后才与比对样本进行比较, 从而避免人工拆分的先入为主和个人偏好。
目前, 国外的几个混合样本拆分软件的拆分结果, 一般都可以直接导入数据库中进行查询比对。而在我国由于一些限制, 从计算拆分得到的数据还无法直接批量导入数据库进行比对; 同时, 大量“ 非真” 数据的导入也会增加比对工作量和随机比中概率。事实上, PCR过程并不是一个完美的理想状态, 能够保证混合样本在扩增前后比例完全保持不变, 而这一点恰恰是目前许多计算拆分工具依赖的最基本前提, 所以今后将PCR扩增过程中的可变性考虑在内也是研究方向之一。
混合样本, 特别是多人混合样本分析仍是一个非常复杂的问题, 目前伴随DNA检验微量化而来的是越来越多的混合分型, 如何通过精确的计算结合有经验的专家对混合样本进行深入分析, 对案情分析和案件侦破都会发挥越来越重要的作用。国外对混合样本的分析非常重视和谨慎, 并且也积极培训鉴定人掌握最新的技术和方法, 这些都值得我们学习和借鉴。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|